
The Possible Mechanism of Amyloid Transformation Based on the Geometrical Parameters of Early-Stage Intermediate in Silico Model for Protein Folding



Abstract
:1. Introduction
- The specificity of the structure of the amyloid form of the polypeptide chain—the fibril component;
- The mechanism of structural transformation leading from the native form of the amyloid (transthyretin used here as an example) chain to its amyloid form using the previously proposed two-step model of protein folding. The analysis presented now mainly uses the early-stage model.
- Our model of early-stage protein folding [32,33], described shortly in the Materials and Methods section as well as in Supplementary Materials–Figures S1 and S3. According to this model, it is assumed that amyloid transformation requires the unfolding treated as return to its early-stage form of folding.
2. Results
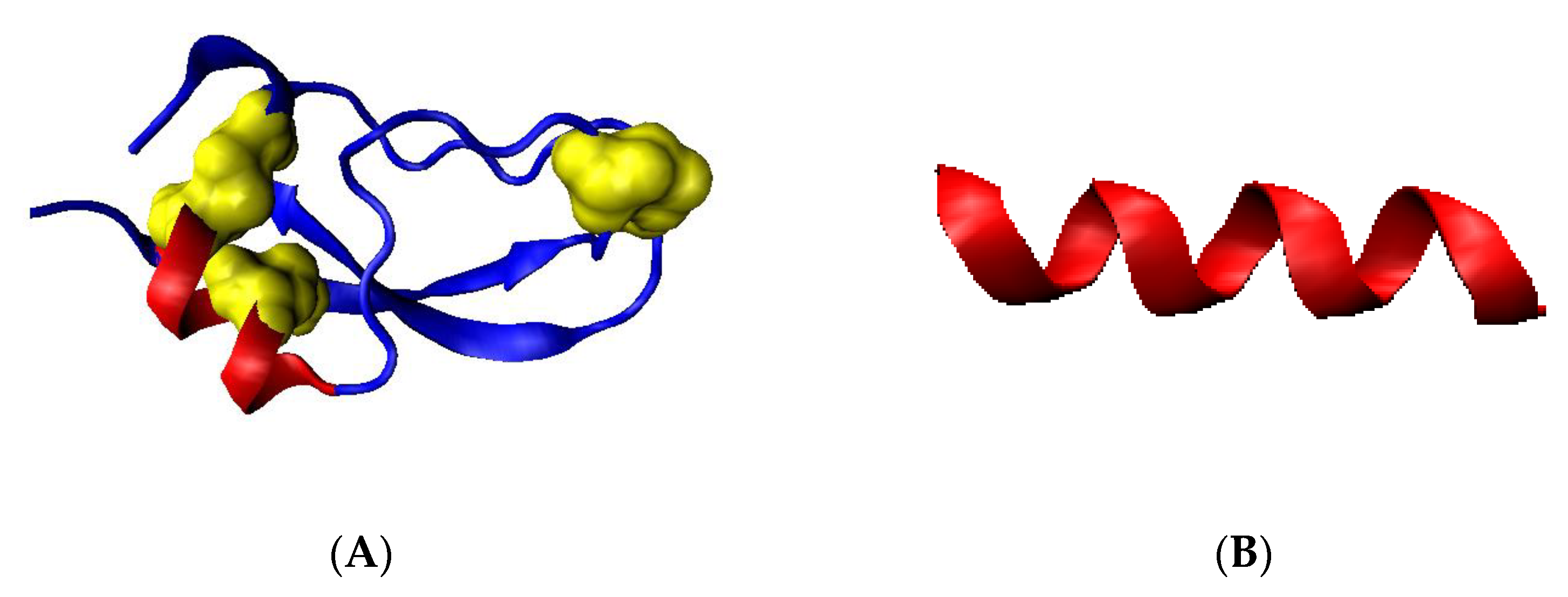
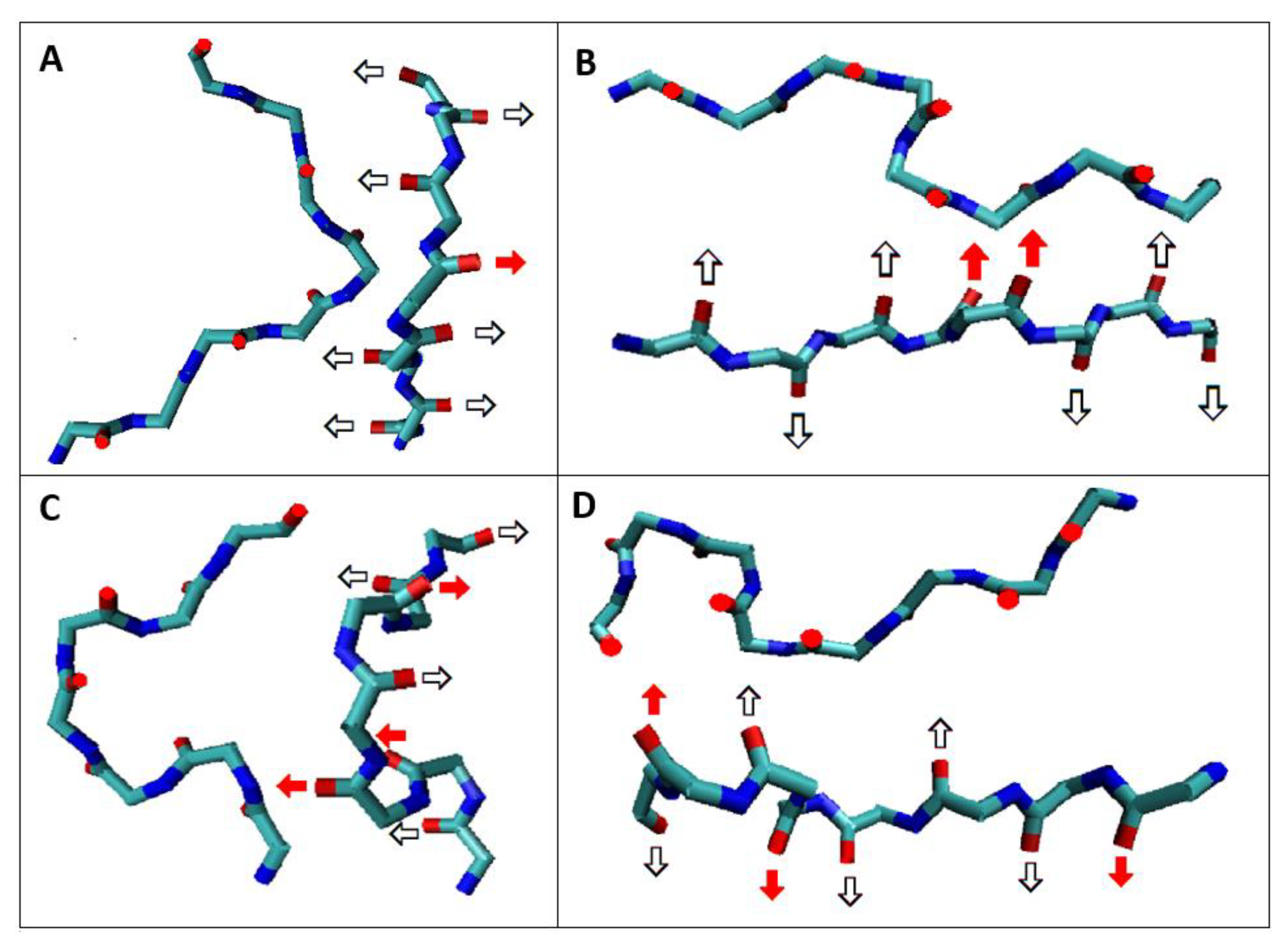
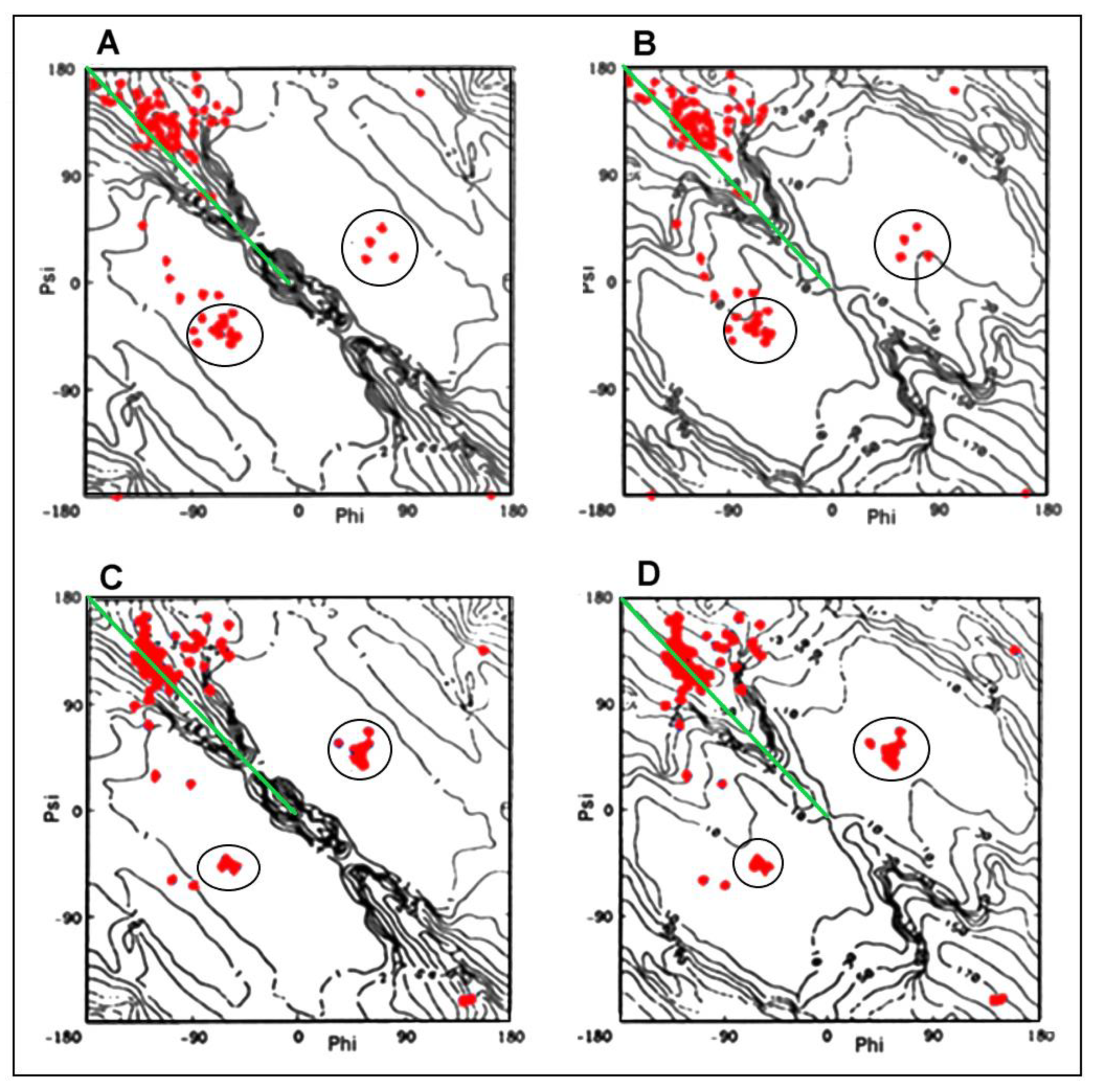
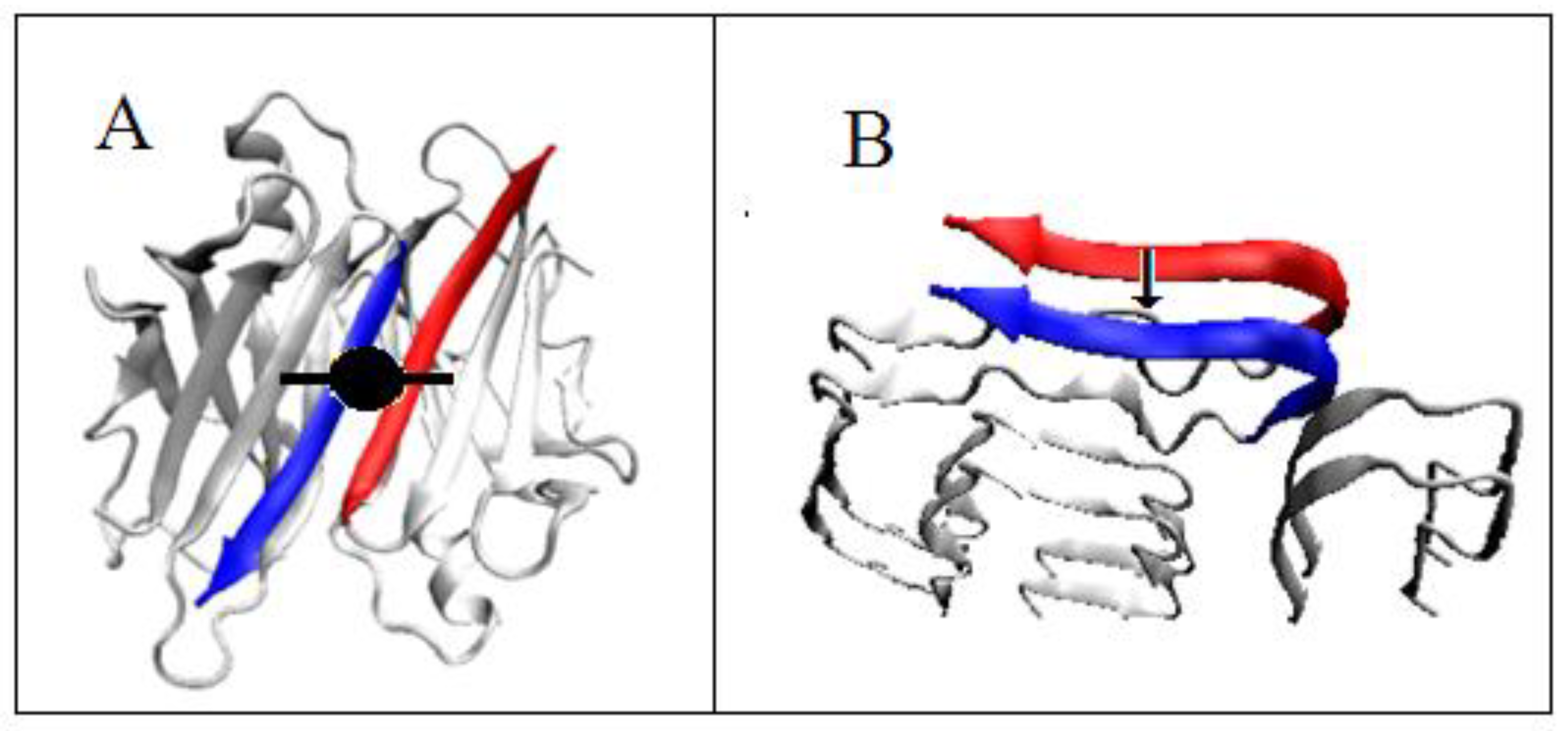
2.1. Analysis of the Specificity of Amyloid Structure Based on Transthyretin (PDB ID—6SDZ)
2.2. The Relation of the Native Form to That Present in the Amyloid Fibril
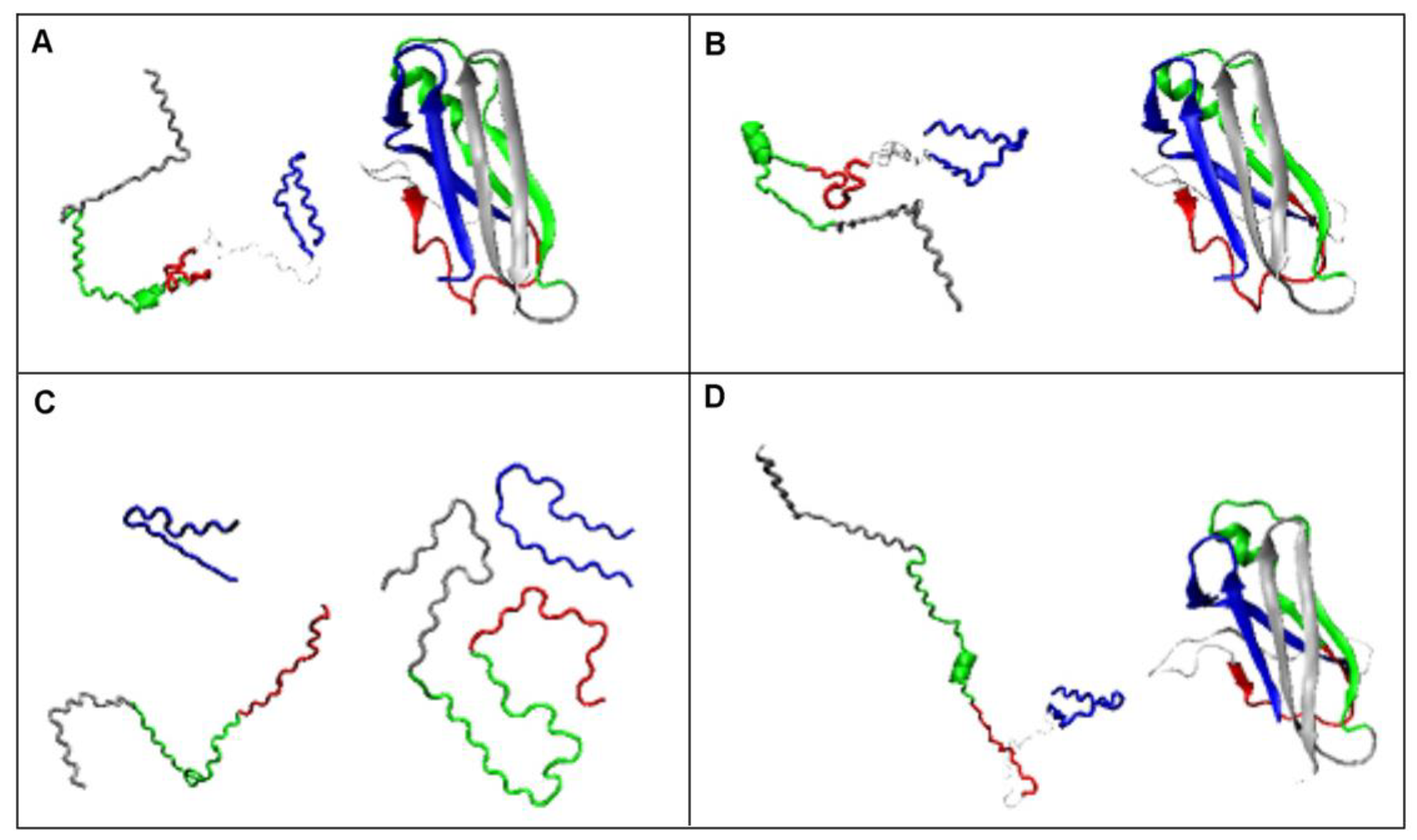
2.2.1. Change of Conformation
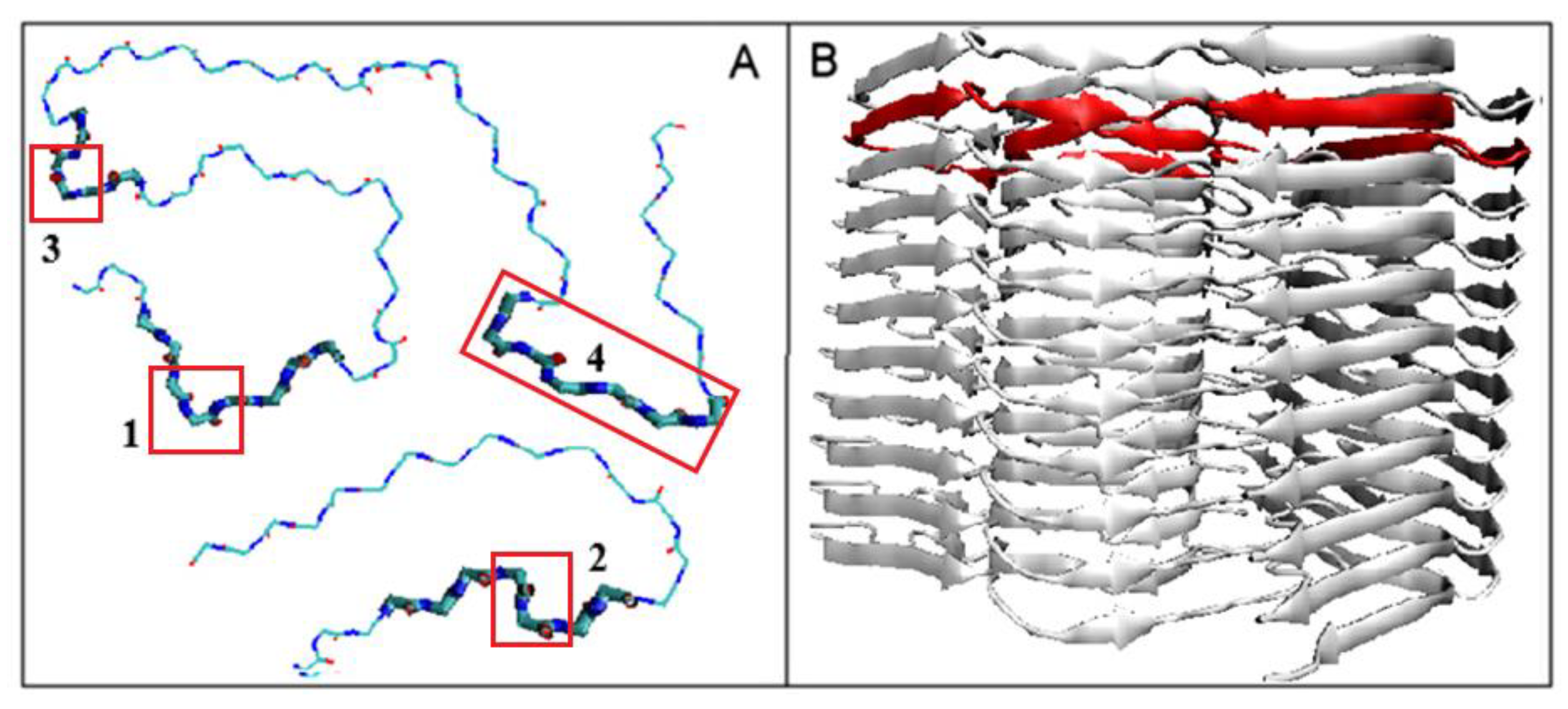
2.2.2. Can the Amyloid Be Called IV-Order Structure?
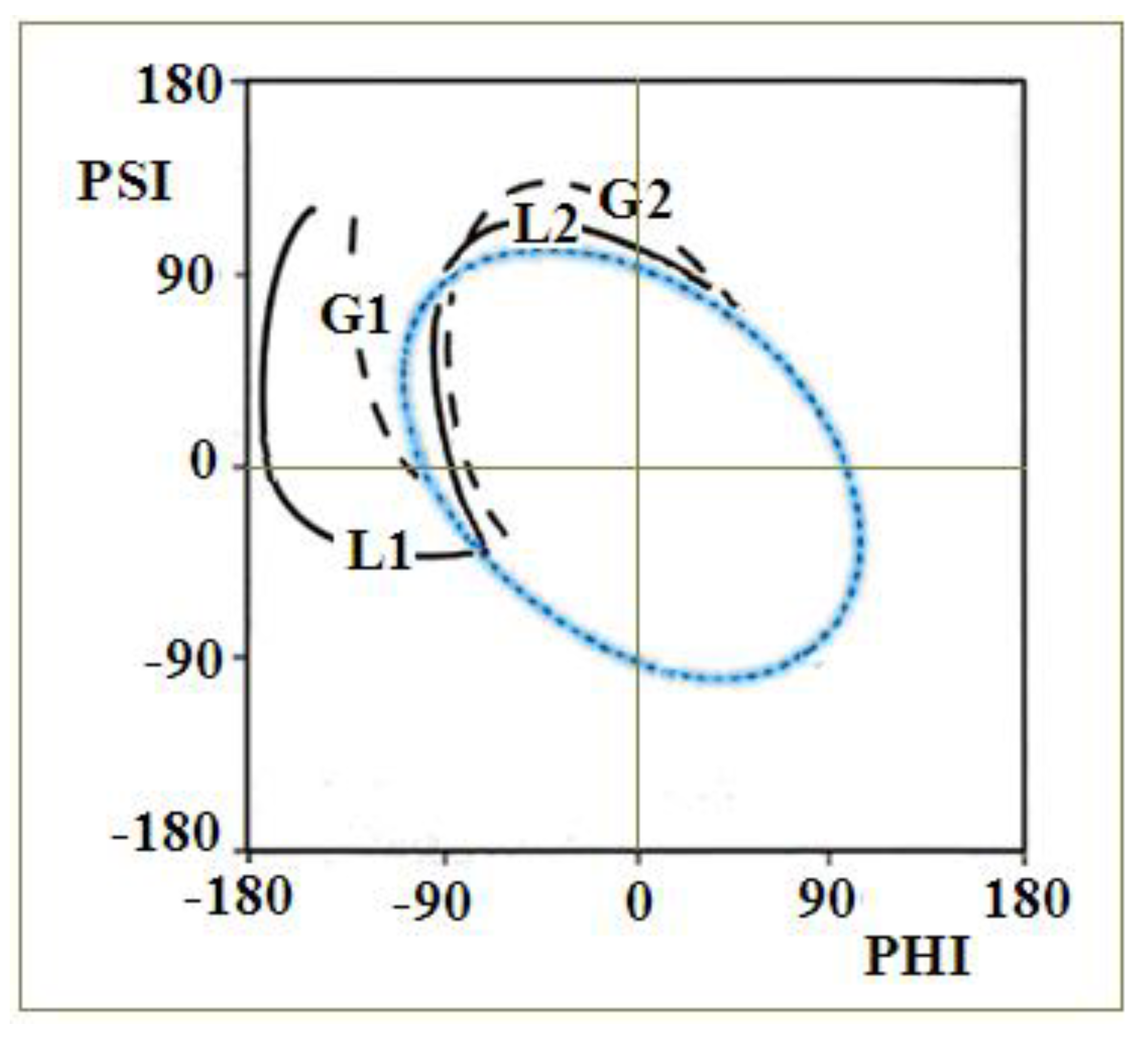
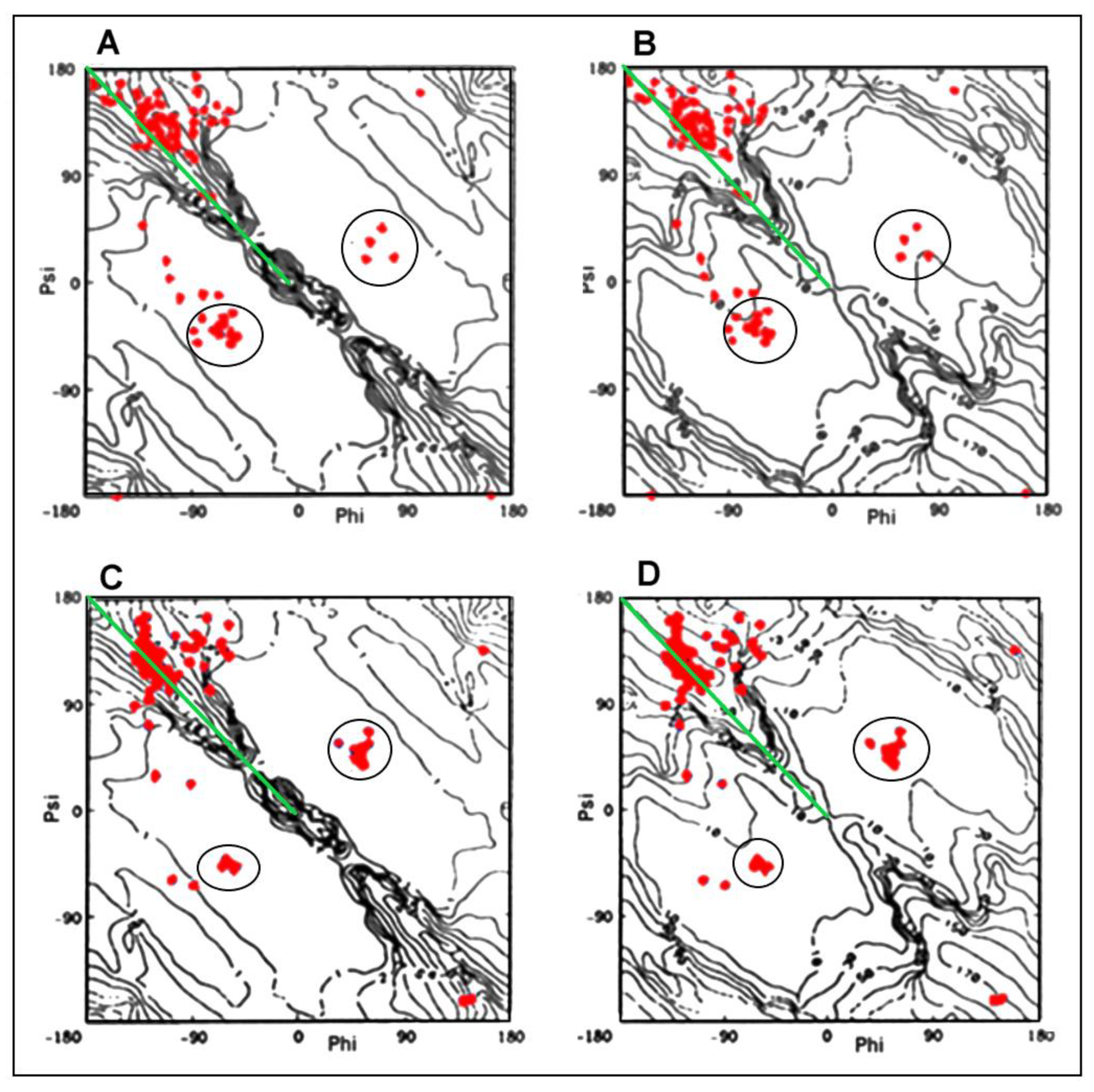
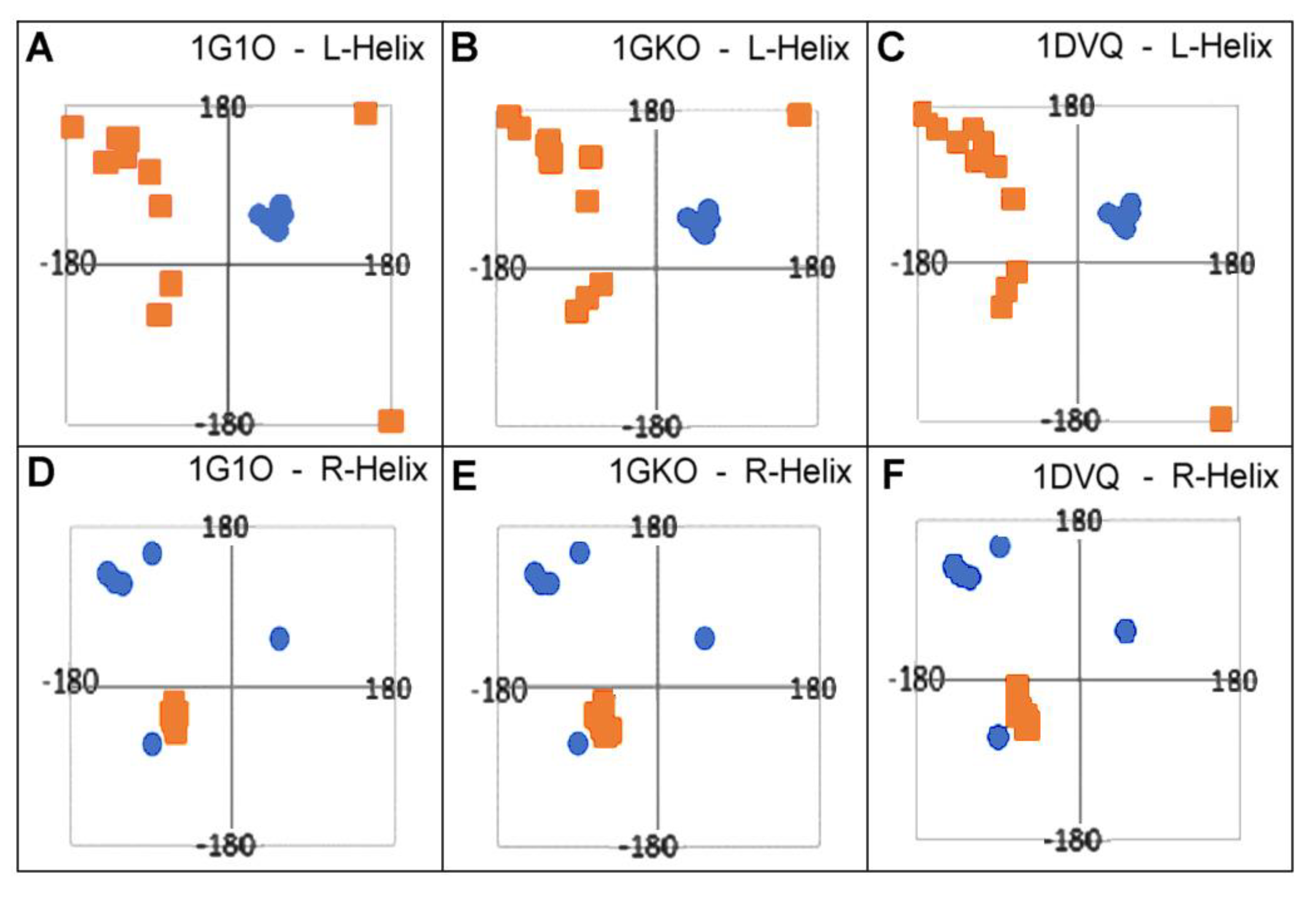
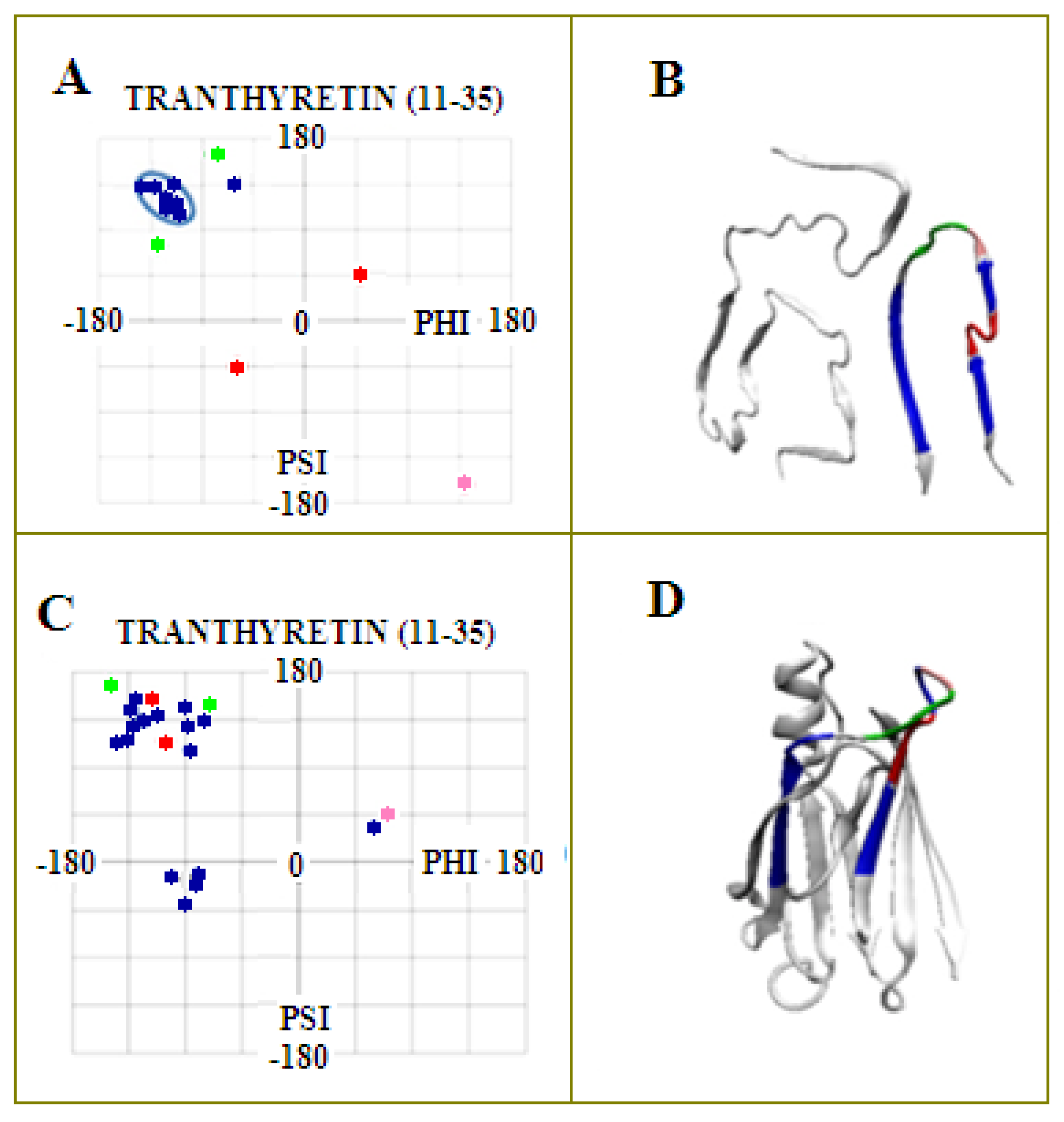
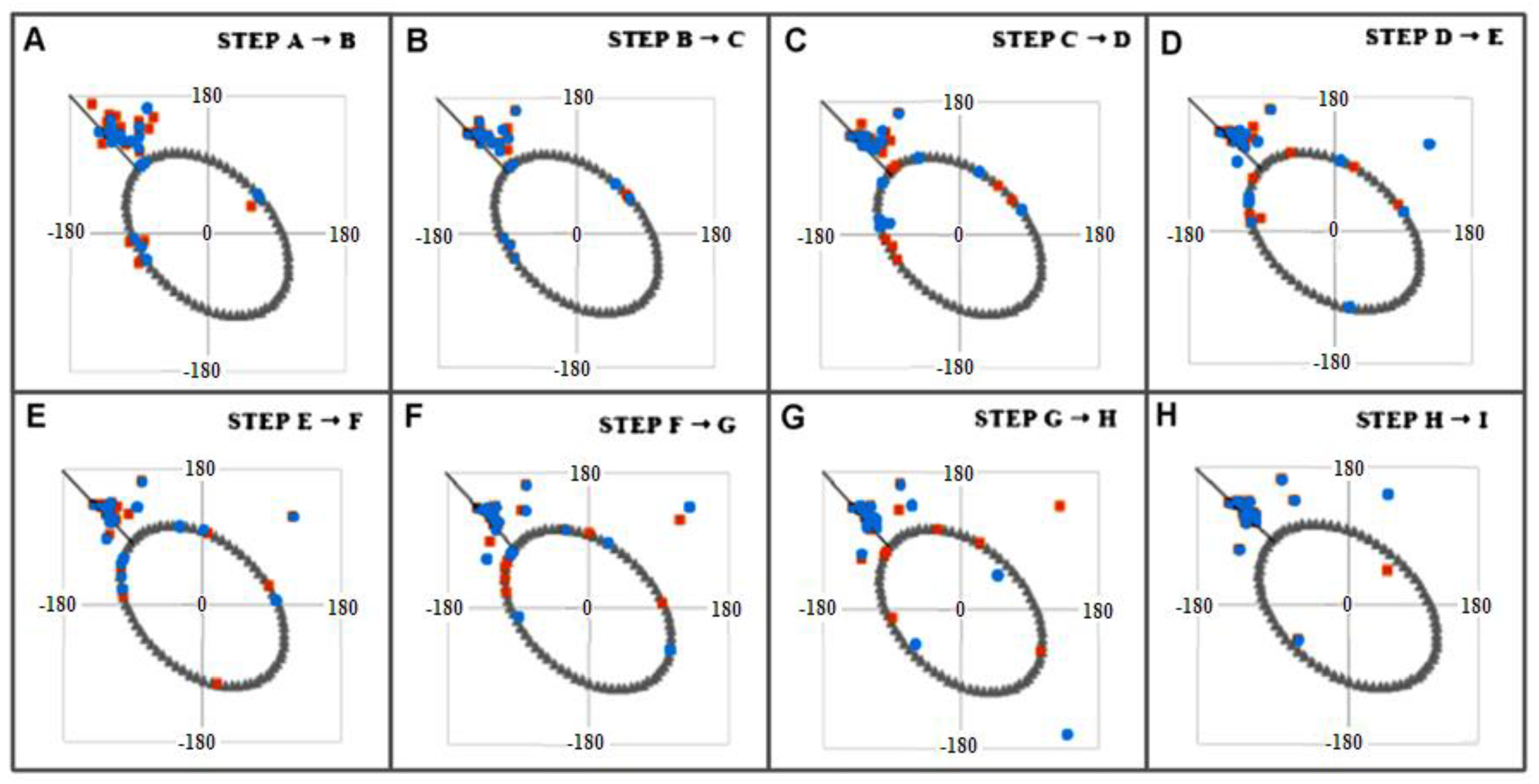
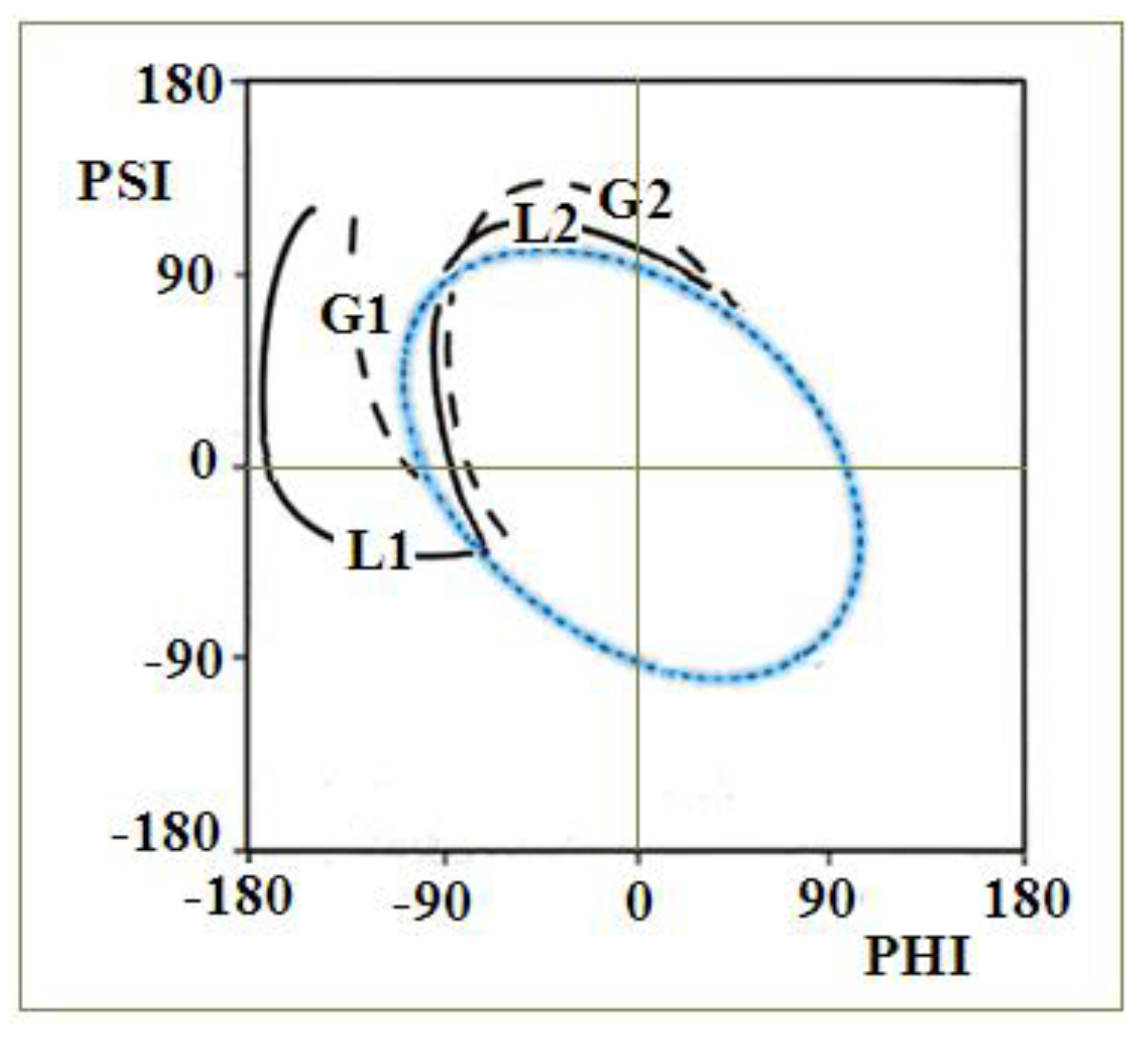
2.3. Modeling the Structural Changes According to the Elliptical Path from the Early-Stage Model of Protein Folding
2.4. The Proposed Model of Conformational Changes Leading to the Amyloid Form of Transthyretin
3. Discussion
4. Materials and Methods
4.1. The Early Stage Model of the Protein Folding Process

4.2. Programs Used
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The protein folding problem. Annu Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J. 2021, 40, 522–544. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.C.; Shrestha, B.; Adhikari, B.; Kc, D.B. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Lupas, A.N.; Pereira, J.; Alva, V.; Merino, F.; Coles, M.; Hartmann, M.D. The breakthrough in protein structure prediction. Biochem. J. 2021, 478, 1885–1890. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, S.Y. Accurate prediction of inter-protein residue-residue contacts for homo-oligomeric protein complexes. Brief. Bioinform. 2021, 22, bbab038. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins 2021, 89, 1607–1617. [Google Scholar] [CrossRef]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins 2021, 89, 1687–1699. [Google Scholar] [CrossRef]
- Guzenko, D.; Lafita, A.; Monastyrskyy, B.; Kryshtafovych, A.; Duarte, J.M. Assessment of protein assembly prediction in CASP13. Proteins 2019, 87, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Hardin, C.; Pogorelov, T.V.; Luthey-Schulten, Z. Ab initio protein structure prediction. Curr. Opin. Struct. Biol. 2002, 12, 176–181. [Google Scholar] [CrossRef]
- Bonneau, R.; Baker, D. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Khalili, M.; Scheraga, H.A. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. USA 2005, 102, 2362–2367. [Google Scholar] [CrossRef] [Green Version]
- Pillardy, J.; Czaplewski, C.; Liwo, A.; Lee, J.; Ripoll, D.R.; Kaźmierkiewicz, R.; Oldziej, S.; Wedemeyer, W.J.; Gibson, K.D.; Arnautova, Y.A.; et al. Recent improvements in prediction of protein structure by global optimization of a potential energy function. Proc. Natl. Acad. Sci. USA 2001, 98, 2329–2333. [Google Scholar] [CrossRef] [Green Version]
- Sieradzan, A.K.; Hansmann, U.H.; Scheraga, H.A.; Liwo, A. Extension of UNRES force field to treat polypeptide chains with D-amino-acid residues. J. Chem. Theory Comput. 2012, 8, 4746–4757. [Google Scholar] [CrossRef]
- Roterman, I.K.; Lambert, M.H.; Gibson, K.D.; Scheraga, H.A. A comparison of the CHARMM, AMBER and ECEPP potentials for peptides. II. Phi-psi maps for N-acetyl alanine N’-methyl amide: Comparisons, contrasts and simple experimental tests. J. Biomol. Struct. Dyn. 1989, 7, 421–453. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.K.; Gibson, K.D.; Scheraga, H.A. A comparison of the CHARMM, AMBER and ECEPP potentials for peptides. I. Conformational predictions for the tandemly repeated peptide (Asn-Ala-Asn-Pro)9. J. Biomol. Struct. Dyn. 1989, 7, 391–419. [Google Scholar] [CrossRef]
- John, B.; Sali, A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003, 31, 3982–3992. [Google Scholar] [CrossRef]
- Martin, A.C.; MacArthur, M.W.; Thornton, J.M. Assessment of comparative modeling in CASP2. Proteins 1997, 29 (Suppl. S1), 14–28. [Google Scholar] [CrossRef]
- Sali, A.; Potterton, L.; Yuan, F.; van Vlijmen, H.; Karplus, M. Evaluation of comparative protein modeling by MODELLER. Proteins 1995, 23, 318–326. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Khoury, G.A.; Liwo, A.; Khatib, F.; Zhou, H.; Chopra, G.; Bacardit, J.; Bortot, L.O.; Faccioli, R.A.; Deng, X.; He, Y.; et al. WeFold: A coopetition for protein structure prediction. Proteins 2014, 82, 1850–1868. [Google Scholar] [CrossRef]
- Keasar, C.; McGuffin, L.J.; Wallner, B.; Chopra, G.; Adhikari, B.; Bhattacharya, D.; Blake, L.; Bortot, L.O.; Cao, R.; Dhanasekaran, B.K.; et al. An analysis and evaluation of the WeFold collaborative for protein structure prediction and its pipelines in CASP11 and CASP12. Sci. Rep. 2018, 8, 9939. [Google Scholar] [CrossRef]
- Makin, O.S.; Sikorski, P.; Serpell, L.C. Diffraction to study protein and peptide assemblies. Curr. Opin. Chem. Biol. 2006, 10, 417–422. [Google Scholar] [CrossRef]
- Tycko, R. Insights into the amyloid folding problem from solid-state NMR. Biochemistry 2003, 42, 3151–3159. [Google Scholar] [CrossRef]
- Der-Sarkissian, A.; Jao, C.C.; Chen, J.; Langen, R. Structural organisation of alpha-synuclein studied by site-directed spin labelling. J. Biol. Chem. 2003, 278, 37530–37535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, S.A.; Langen, R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 2004, 279, 48420–48425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Török, M.; Milton, S.; Kayed, R.; Wu, P.; McIntire, T.; Glabe, C.G.; Langen, R. Structural and dynamic features of Alzheimer’s Abeta peptide in amyloid fibrils studied by site-directed spin labelling. J. Biol. Chem. 2002, 277, 40810–40815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef]
- Roterman, I. Modelling the optimal simulation path in the peptide chain folding--studies based on geometry of alanine heptapeptide. J. Biol. 1995, 177, 283–288. [Google Scholar] [CrossRef]
- Roterman, I. The geometrical analysis of peptide backbone structure and its local deformations. Biochimie 1995, 77, 204–216. [Google Scholar] [CrossRef]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [CrossRef]
- Eneqvist, T.; Andersson, K.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, A.E. The beta-slip: A novel concept in transthyretin amyloidosis. Mol. Cell 2000, 6, 1207–1218. [Google Scholar] [CrossRef]
- Jiang, X.; Smith, C.S.; Petrassi, H.M.; Hammarström, P.; White, J.T.; Sacchettini, J.C.; Kelly, J.W. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 2001, 40, 11442–11452. [Google Scholar] [CrossRef]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef] [Green Version]
- Daggett, V.; Levitt, M. Molecular dynamics simulations of helix denaturation. J. Mol. Biol. 1992, 223, 1121–1138. [Google Scholar] [CrossRef]
- Daggett, V.; Levitt, M. Protein unfolding pathways explored through molecular dynamics simulations. J. Mol. Biol. 1993, 232, 600–619. [Google Scholar] [CrossRef] [Green Version]
- Deisenhofer, J.; Steigemann, W. Crystallographic refinement of the structure of Bovine Pancreatic Inhibitor at 1.5 Å resolution. Acta Cryst. 1975, B31, 238–250. [Google Scholar] [CrossRef]
- Dickerson, R.E.; Geis, I. Struktur and Funktion der Proteine; Verlag Chemie: Weinheim, Germany, 1971; p. 26. (In German) [Google Scholar]
- Ghélis, C.; Yon, J. Protein Folding; Academic Press: Cambridge, MA, USA, 1982; p. 51. [Google Scholar] [CrossRef]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model. Int. J. Mol. Sci. 2021, 22, 10587. [Google Scholar] [CrossRef]
- Banach, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Divergence Entropy-Based Evaluation of Hydrophobic Core in Aggressive and Resistant Forms of Transthyretin. Entropy 2021, 23, 458. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi Nussinov, R.; Ishii, Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [Green Version]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed. 2015, 54, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Sgourakis, N.G.; Yau, W.-M.; Qiang, W. Modeling an in-register, parallel “iowa” aβ fibril structure using solid-state NMR data from labeled samples with rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 2011, 411, 312–316. [Google Scholar] [CrossRef]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Kelly, J.W. The environmental dependency of protein folding best explains prion and amyloid diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 930–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim Biophys Acta. 2000, 1502, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Fraser, P.E. Prions and prion-like proteins. J. Biol. Chem. 2014, 289, 19839–19840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Radford, H.; Mallucci, G.R. Prions: Generation and spread versus neurotoxicity. J. Biol. Chem. 2014, 289, 19862–19868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, J.S. Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Creutzfeldt-Jakob disease and scrapie prions. Alzheimer Dis. Assoc. Disord. 1989, 3, 52–78. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. Alternative conformations of amyloidogenic proteins govern their behavior. Curr. Opin. Struct. Biol. 1996, 6, 11–17. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology and pathogenesis of prion diseases. Trends Biochem. Sci. 1996, 21, 482–487. [Google Scholar] [CrossRef]
- Huang, Z.; Prusiner, S.B.; Cohen, F.E. Structures of prion proteins and conformational models for prion diseases. Curr. Top. Microbiol. Immunol. 1996, 207, 49–67. [Google Scholar] [CrossRef]
- Caughey, B.; Kocisko, D.A.; Raymond, G.J.; Lansbury, P.T. Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem. Biol. 1995, 2, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Safar, J. The folding intermediate concept of prion protein formation and conformational links to infectivity. Microbiol. Immunol. 1996, 207, 69–76. [Google Scholar] [CrossRef]
- McParland, V.J.; Kad, N.M.; Kalverda, A.P.; Brown, A.; Kirwin-Jones, P.; Hunter, M.G.; Sunde, M.; Radford, S.E. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry 2000, 39, 8735–8746. [Google Scholar] [CrossRef]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef]
- Eakin, C.M.; Berman, A.J.; Miranker, A.D. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 2006, 13, 202–208. [Google Scholar] [CrossRef]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Stapor, K.; Gądek, K.; Gubała, T.; Nowakowski, P.; Fabian, P.; Konieczny, L. Dependence of Protein Structure on Environment: FOD Model Applied to Membrane Proteins. Membranes 2021, 12, 50. [Google Scholar] [CrossRef]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef]
- Hill, A.F.; Collinge, J. Subclinical prion infection in humans and animals. Br. Med. Bull. 2003, 66, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Andre, R.; Tabrizi, S.J. Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 2012, 6, 32–36. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Guo, J.; Wetzel, R.; Xu, Y. Molecular modeling of the core of Abeta amyloid fibrils. Proteins 2004, 57, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Come, J.; Fraser, P.; Lansbury, P.J. A Kinetic Model for Amyloid Formation In the Prion Diseases, Importance of Seeding. Proc. Natl. Acad. Sci. USA 1993, 90, 5959–5963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perutz, M.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine Repeats as Polar Zippers: Their Possible Role in Inherited Neurodegenerative Diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef] [Green Version]
- Schlunegger, M.P.; Bennett, M.J.; Eisenberg, D. Oligomer formation by 3D domain swapping: A model for protein assembly and misassembly. Adv. Protein Chem. 1997, 50, 61–122. [Google Scholar] [CrossRef]
- Staniforth, R.A.; Giannini, S.; Higgins, L.D.; Conroy, M.J.; Hounslow, A.M.; Jerala, R.; Craven, C.J.; Waltho, J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001, 20, 4774–4781. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative disease. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Zibaee, S.; Makin, O.S.; Goedert, M.; Serpell, L.C. A simple algorithm locates beta-strands in the amyloid fibril core of alpha-synuclein, Abeta and tau using the amino acid sequence alone. Protein Sci. 2007, 16, 906–918. [Google Scholar] [CrossRef] [Green Version]
- Karamanos, T.K.; Kalverda, A.P.; Radford, S.E. Generating Ensembles of Dynamic Misfolding Proteins. Front. Neurosci. 2022, 16, 881534. [Google Scholar] [CrossRef]
- Geddes, A.J.; Parker, K.D.; Atkins, E.D.T.; Beighton, E. “Cross β” Conformation in Protein. J. Mol. Biol. 1968, 32, 343–358. [Google Scholar] [CrossRef]
- Brylinski, M.; Konieczny, L.; Czerwonko, P.; Jurkowski, W.; Roterman, I. Early-stage folding in proteins (in silico) sequence-to-structure relations. J. Biomed. Biotechnol. 2005, 2005, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Fabian, P.; Stapor, K.; Roterman, I. Model of Early Stage Intermediate in Respect to Its Final Structure. Biomolecules 2019, 9, 866. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, R.; Ranson, N.A.; Radford, S.E. Amyloid structures: Much more than just a cross-beta fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. [Google Scholar] [CrossRef]
- Harpham, M.R.; Levinger, N.E.; Ladanyi, B.M. An investigation of water dynamics in binary mixtures of water and dimethyl sulfoxide. J. Phys. Chem. B 2008, 112, 283–293. [Google Scholar] [CrossRef]
- Laage, D.; Hynes, J.T. A molecular jump mechanism of water reorientation. Science 2006, 311, 832–835. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Ma, C.D.; Wang, C.; Acevedo-Vélez, C.; Gellman, S.H.; Abbott, N.L. Modulation of hydrophobic interactions by proximally immobilized ions. Nature 2015, 517, 347–350. [Google Scholar] [CrossRef]
- Macias-Romero, C.; Nahalka, I.; Okur, H.I.; Roke, S. Optical imaging of surface chemistry and dynamics in confinement. Science 2017, 357, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Gallo, P.; Stanley, H.E. Supercooled water reveals its secrets. Science 2017, 358, 1543–1544. [Google Scholar] [CrossRef]
- Gallo, P.; Amann-Winkel, K.; Angell, C.A.; Anisimov, M.A.; Caupin, F.; Chakravarty, C.; Lascaris, E.; Loerting, T.; Panagiotopoulos, A.Z.; Russo, J.; et al. Water: A Tale of Two Liquids. Chem. Rev. 2016, 116, 7463–7500. [Google Scholar] [CrossRef]
- Corradini, D.; Strekalova, E.G.; Stanley, H.E.; Gallo, P. Microscopic mechanism of protein cryopreservation in an aqueous solution with trehalose. Sci. Rep. 2013, 3, 1218. [Google Scholar] [CrossRef]
- Corradini, D.; Su, Z.; Stanley, H.E.; Gallo, P. A molecular dynamics study of the equation of state and the structure of supercooled aqueous solutions of methanol. J. Chem. Phys. 2012, 137, 184503. [Google Scholar] [CrossRef] [Green Version]
- Strekalova, E.G.; Corradini, D.; Mazza, M.G.; Buldyrev, S.V.; Gallo, P.; Franzese, G.; Stanley, H.E. Effect of hydrophobic environments on the hypothesized liquid-liquid critical point of water. J. Biol. Phys. 2012, 38, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Biancalana, M.; Makabe, K.; Koide, S. Minimalist design of water-soluble cross-beta architecture. Proc. Natl. Acad. Sci. USA 2010, 107, 3469–3474. [Google Scholar] [CrossRef] [Green Version]
- Corradini, D.; Gallo, P.; Buldyrev, S.V.; Stanley, H.E. Fragile-to-strong crossover coupled to the liquid-liquid transition in hydrophobic solutions. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012, 85 Pt 1, 051503. [Google Scholar] [CrossRef] [Green Version]
- Corradini, D.; Buldyrev, S.V.; Gallo, P.; Stanley, H.E. Effect of hydrophobic solutes on the liquid-liquid critical point. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010, 81 Pt 1, 061504. [Google Scholar] [CrossRef] [Green Version]
- Clary, D.C. Quantum dynamics in the smallest water droplet. Science 2016, 351, 1267–1268. [Google Scholar] [CrossRef]
- Richardson, J.O.; Pérez, C.; Lobsiger, S.; Reid, A.A.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Wales, D.J.; Pate, B.H.; Althorpe, S.C. Concerted hydrogen-bond breaking by quantum tunneling in the water hexamer prism. Science 2016, 351, 1310–1333. [Google Scholar] [CrossRef] [Green Version]
- Lupi, L.; Hudait, A.; Peters, B.; Grünwald, M.; Gotchy Mullen, R.; Nguyen, A.H.; Molinero, V. Role of stacking disorder in ice nucleation. Nature 2017, 551, 218–222. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NR | Codes of Specific Conformation | 3D Structure of Conformation |
|---|---|---|
| 1 | AB-63L-AB | 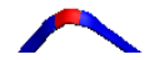 |
| 2 | AB-17L-18R-AB | 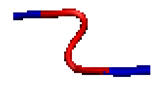 |
| 3 | AB-87L-88R-88B-89L-90B-AB |  |
| 4 | AB-105M-106L-107B-108AB-109L-110B-111L-112B-113AB |  |
| R-α-Helix | L-α-Helix | β-Structural Area | |||
|---|---|---|---|---|---|
| E + F | E | F | |||
| 6SDZ | 4.84 | 7.80 | 14.77 (64) | 9.65 (56) | 50.60 (8) |
| 1DVQ | 24.05 | 12.10 | 19.95 (70) | 13.18 (58) | 52.67 (12) |
| 1GKO | 35.18 | 14.91 | 19.37 (74) | 13.43 (62) | 50.08 (12) |
| 1G1O | 14.80 | 13.27 | 20.98 (78) | 13.64 (64) | 54.56 (14) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roterman, I.; Stapor, K.; Dułak, D.; Konieczny, L. The Possible Mechanism of Amyloid Transformation Based on the Geometrical Parameters of Early-Stage Intermediate in Silico Model for Protein Folding. Int. J. Mol. Sci. 2022, 23, 9502. https://doi.org/10.3390/ijms23169502
Roterman I, Stapor K, Dułak D, Konieczny L. The Possible Mechanism of Amyloid Transformation Based on the Geometrical Parameters of Early-Stage Intermediate in Silico Model for Protein Folding. International Journal of Molecular Sciences. 2022; 23(16):9502. https://doi.org/10.3390/ijms23169502
Chicago/Turabian StyleRoterman, Irena, Katarzyna Stapor, Dawid Dułak, and Leszek Konieczny. 2022. "The Possible Mechanism of Amyloid Transformation Based on the Geometrical Parameters of Early-Stage Intermediate in Silico Model for Protein Folding" International Journal of Molecular Sciences 23, no. 16: 9502. https://doi.org/10.3390/ijms23169502
APA StyleRoterman, I., Stapor, K., Dułak, D., & Konieczny, L. (2022). The Possible Mechanism of Amyloid Transformation Based on the Geometrical Parameters of Early-Stage Intermediate in Silico Model for Protein Folding. International Journal of Molecular Sciences, 23(16), 9502. https://doi.org/10.3390/ijms23169502