

Neuronal Guidance Molecules in Bone Remodeling and Orthodontic Tooth Movement


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bone Remodeling
2.1. Neuronal Guidance Molecules in Bone Remodeling
2.1.1. Netrins
2.1.2. Ephrins and Eph Receptors
2.1.3. Semaphorins
2.1.4. Slits
2.2. Neuronal Guidance Molecules in Orthodontic Tooth Movement
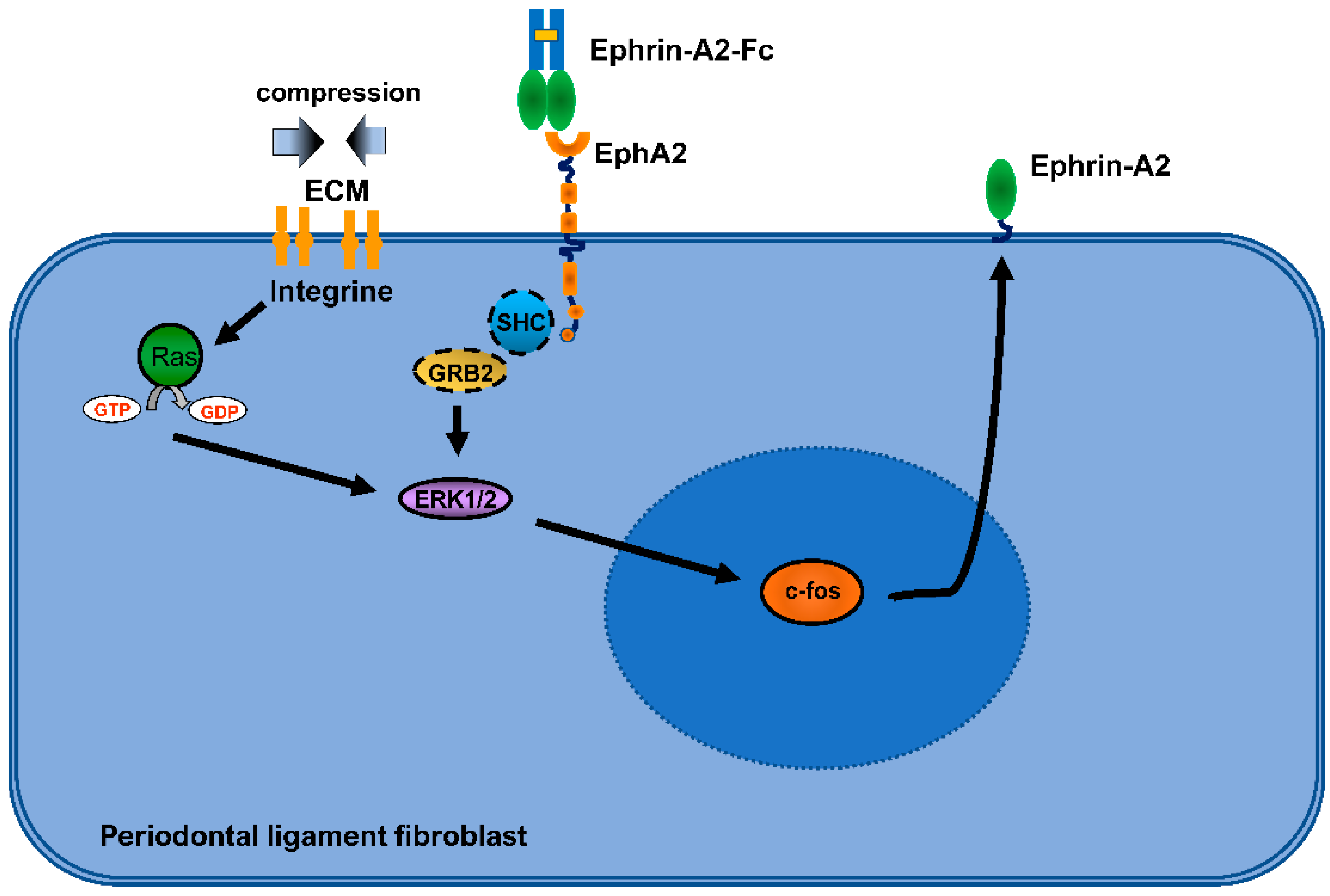
2.2.1. Ephrins and Eph Receptors
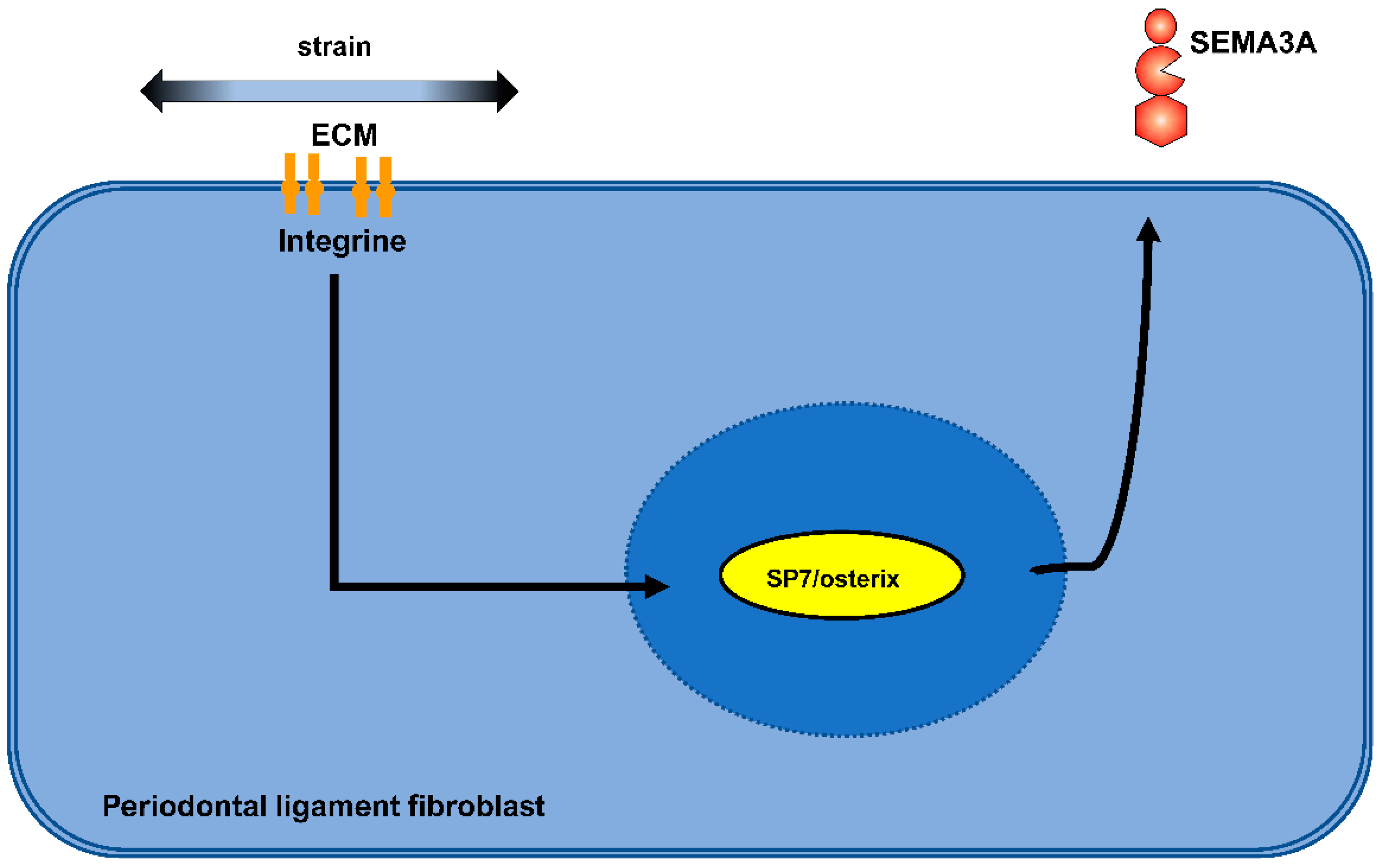
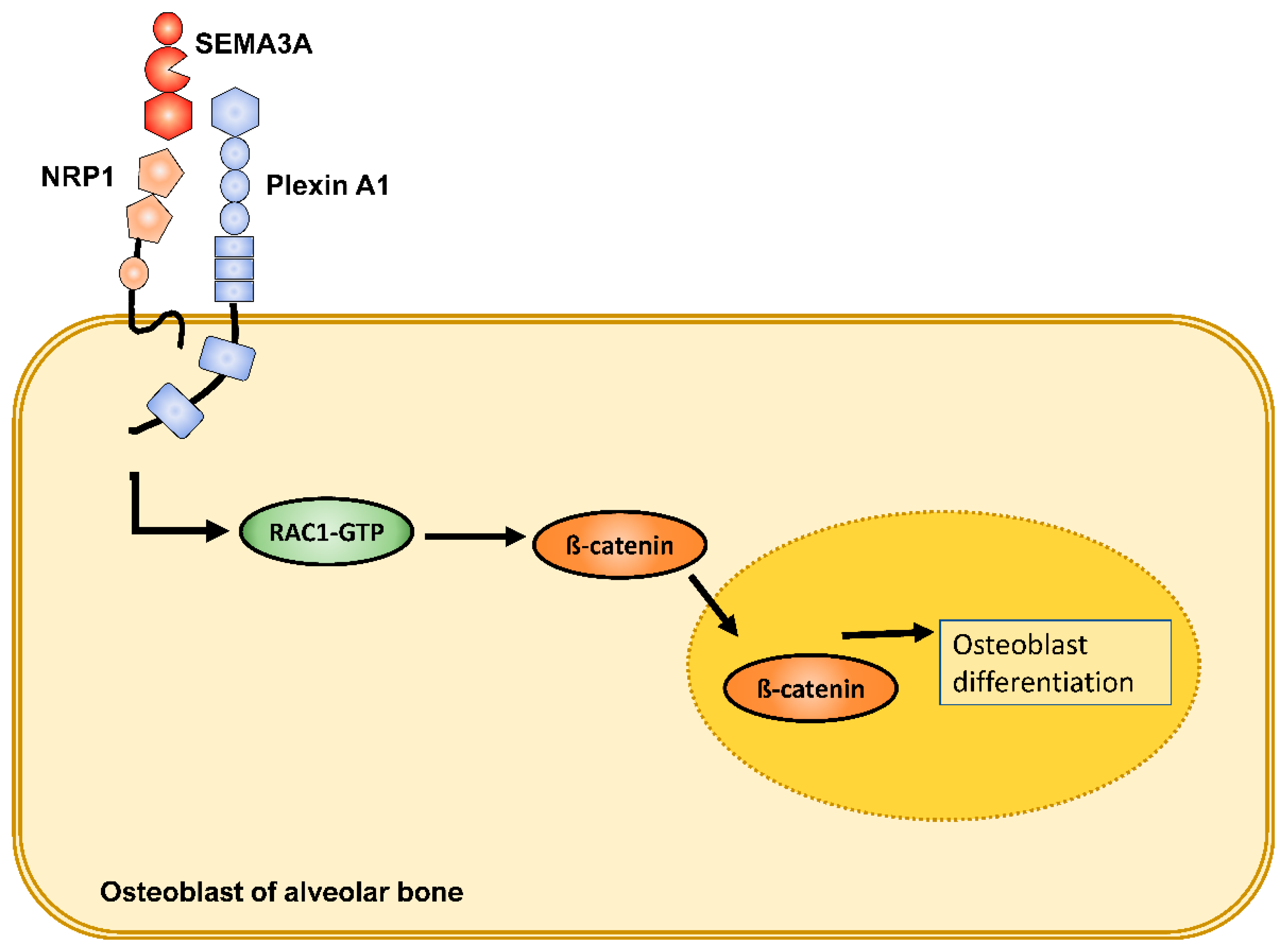
2.2.2. Semaphorins
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krishnan, V.; Davidovitch, Z. On a path to unfolding the biological mechanisms of orthodontic tooth movement. J. Dent. Res. 2009, 88, 597–608. [Google Scholar] [CrossRef]
- Masella, R.S.; Meister, M. Current concepts in the biology of orthodontic tooth movement. Am. J. Orthod. Dentofacial Orthop. 2006, 129, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Henneman, S.; Von den Hoff, J.W.; Maltha, J.C. Mechanobiology of tooth movement. Eur. J. Orthod 2008, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Wise, G.E.; King, G.J. Mechanisms of tooth eruption and orthodontic tooth movement. J. Dent. Res. 2008, 87, 414–434. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A. Bone mass homeostasis and bisphosphonate action. Bone 1997, 20, 1–4. [Google Scholar] [CrossRef]
- Ehrlich, P.J.; Lanyon, L.E. Mechanical strain and bone cell function: A review. Osteoporos. Int. 2002, 13, 688–700. [Google Scholar] [CrossRef]
- Vansant, L.; Cadenas De Llano-Perula, M.; Verdonck, A.; Willems, G. Expression of biological mediators during orthodontic tooth movement: A systematic review. Arch. Oral. Biol. 2018, 95, 170–186. [Google Scholar] [CrossRef]
- Hinck, L. The versatile roles of “axon guidance” cues in tissue morphogenesis. Dev. Cell 2004, 7, 783–793. [Google Scholar] [CrossRef]
- Abeynayake, N.; Arthur, A.; Gronthos, S. Crosstalk between skeletal and neural tissues is critical for skeletal health. Bone 2021, 142, 115645. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Kang, S.; Kumanogoh, A. Crosstalk between axon guidance signaling and bone remodeling. Bone 2022, 157, 116305. [Google Scholar] [CrossRef]
- Luukko, K.; Kettunen, P. Integration of tooth morphogenesis and innervation by local tissue interactions, signaling networks, and semaphorin 3A. Cell Adh. Migr. 2016, 10, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Loes, S.; Luukko, K.; Kvinnsland, I.H.; Kettunen, P. Slit1 is specifically expressed in the primary and secondary enamel knots during molar tooth cusp formation. Mech. Dev. 2001, 107, 155–157. [Google Scholar] [CrossRef]
- Loes, S.; Kettunen, P.; Kvinnsland, I.H.; Taniguchi, M.; Fujisawa, H.; Luukko, K. Expression of class 3 semaphorins and neuropilin receptors in the developing mouse tooth. Mech. Dev. 2001, 101, 191–194. [Google Scholar] [CrossRef]
- Luukko, K.; Loes, S.; Kvinnsland, I.H.; Kettunen, P. Expression of ephrin-A ligands and EphA receptors in the developing mouse tooth and its supporting tissues. Cell Tissue Res. 2005, 319, 143–152. [Google Scholar] [CrossRef]
- Crockett, J.C.; Rogers, M.J.; Coxon, F.P.; Hocking, L.J.; Helfrich, M.H. Bone remodelling at a glance. J. Cell Sci. 2011, 124, 991–998. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar] [CrossRef]
- Koga, T.; Inui, M.; Inoue, K.; Kim, S.; Suematsu, A.; Kobayashi, E.; Iwata, T.; Ohnishi, H.; Matozaki, T.; Kodama, T.; et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 2004, 428, 758–763. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef]
- Culotti, J.G.; Merz, D.C. DCC and netrins. Curr. Opin. Cell Biol. 1998, 10, 609–613. [Google Scholar] [CrossRef]
- Cole, S.J.; Bradford, D.; Cooper, H.M. Neogenin: A multi-functional receptor regulating diverse developmental processes. Int. J. Biochem. Cell Biol. 2007, 39, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Bradford, D.; Cole, S.J.; Cooper, H.M. Netrin-1: Diversity in development. Int. J. Biochem. Cell Biol. 2009, 41, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Van Vactor, D.; Flanagan, J.G. The middle and the end: Slit brings guidance and branching together in axon pathway selection. Neuron 1999, 22, 649–652. [Google Scholar] [CrossRef]
- Raper, J.A. Semaphorins and their receptors in vertebrates and invertebrates. Curr. Opin. Neurobiol. 2000, 10, 88–94. [Google Scholar] [CrossRef]
- Wilkinson, D.G. Eph receptors and ephrins: Regulators of guidance and assembly. Int. Rev. Cytol. 2000, 196, 177–244. [Google Scholar] [PubMed]
- Wilkinson, D.G. Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci. 2001, 2, 155–164. [Google Scholar] [CrossRef]
- Rajasekharan, S.; Kennedy, T.E. The netrin protein family. Genome Biol. 2009, 10, 239. [Google Scholar] [CrossRef]
- Liu, Y.; Stein, E.; Oliver, T.; Li, Y.; Brunken, W.J.; Koch, M.; Tessier-Lavigne, M.; Hogan, B.L. Novel role for Netrins in regulating epithelial behavior during lung branching morphogenesis. Curr Biol. 2004, 14, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Ly, N.P.; Komatsuzaki, K.; Fraser, I.P.; Tseng, A.A.; Prodhan, P.; Moore, K.J.; Kinane, T.B. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 14729–14734. [Google Scholar] [CrossRef]
- Causeret, F.; Hidalgo-Sanchez, M.; Fort, P.; Backer, S.; Popoff, M.R.; Gauthier-Rouviere, C.; Bloch-Gallego, E. Distinct roles of Rac1/Cdc42 and Rho/Rock for axon outgrowth and nucleokinesis of precerebellar neurons toward netrin 1. Development 2004, 131, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Forcet, C.; Stein, E.; Pays, L.; Corset, V.; Llambi, F.; Tessier-Lavigne, M.; Mehlen, P. Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 2002, 417, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; Hoshino, A.; Tsai, L.; Henley, J.R.; Goshima, Y.; Tessier-Lavigne, M.; Poo, M.M.; Hong, K. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 2003, 423, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Mediero, A.; Ramkhelawon, B.; Perez-Aso, M.; Moore, K.J.; Cronstein, B.N. Netrin-1 is a critical autocrine/paracrine factor for osteoclast differentiation. J. Bone Miner. Res. 2015, 30, 837–854. [Google Scholar] [CrossRef]
- Zhu, S.; Zhu, J.; Zhen, G.; Hu, Y.; An, S.; Li, Y.; Zheng, Q.; Chen, Z.; Yang, Y.; Wan, M.; et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J. Clin. Invest. 2019, 129, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Eichmann, A. Axon guidance molecules in vascular patterning. Cold Spring Harb. Perspect. Biol. 2010, 2, a001875. [Google Scholar] [CrossRef]
- Enoki, Y.; Sato, T.; Tanaka, S.; Iwata, T.; Usui, M.; Takeda, S.; Kokabu, S.; Matsumoto, M.; Okubo, M.; Nakashima, K.; et al. Netrin-4 derived from murine vascular endothelial cells inhibits osteoclast differentiation in vitro and prevents bone loss in vivo. FEBS Lett. 2014, 588, 2262–2269. [Google Scholar] [CrossRef]
- Enoki, Y.; Sato, T.; Kokabu, S.; Hayashi, N.; Iwata, T.; Yamato, M.; Usui, M.; Matsumoto, M.; Tomoda, T.; Ariyoshi, W.; et al. Netrin-4 Promotes Differentiation and Migration of Osteoblasts. In Vivo 2017, 31, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Jernvall, J.; Thesleff, I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech. Dev. 2000, 92, 19–29. [Google Scholar] [CrossRef]
- Mellott, D.O.; Burke, R.D. The molecular phylogeny of eph receptors and ephrin ligands. BMC Cell Biol. 2008, 9, 27. [Google Scholar] [CrossRef]
- Eph Nomenclature Committee. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 1997, 90, 403–404. [Google Scholar] [CrossRef] [Green Version]
- Gale, N.W.; Holland, S.J.; Valenzuela, D.M.; Flenniken, A.; Pan, L.; Ryan, T.E.; Henkemeyer, M.; Strebhardt, K.; Hirai, H.; Wilkinson, D.G.; et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 1996, 17, 9–19. [Google Scholar] [CrossRef]
- Pasquale, E.B. The Eph family of receptors. Curr. Opin. Cell Biol. 1997, 9, 608–615. [Google Scholar] [CrossRef]
- Bartley, T.D.; Hunt, R.W.; Welcher, A.A.; Boyle, W.J.; Parker, V.P.; Lindberg, R.A.; Lu, H.S.; Colombero, A.M.; Elliott, R.L.; Guthrie, B.A.; et al. B61 is a ligand for the ECK receptor protein-tyrosine kinase. Nature 1994, 368, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol 2005, 6, 462–475. [Google Scholar] [CrossRef]
- Gerety, S.S.; Wang, H.U.; Chen, Z.F.; Anderson, D.J. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 1999, 4, 403–414. [Google Scholar] [CrossRef]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef]
- Arthur, A.; Panagopoulos, R.A.; Cooper, L.; Menicanin, D.; Parkinson, I.H.; Codrington, J.D.; Vandyke, K.; Zannettino, A.C.; Koblar, S.A.; Sims, N.A.; et al. EphB4 enhances the process of endochondral ossification and inhibits remodeling during bone fracture repair. J. Bone Miner. Res. 2013, 28, 926–935. [Google Scholar] [CrossRef]
- Irie, N.; Takada, Y.; Watanabe, Y.; Matsuzaki, Y.; Naruse, C.; Asano, M.; Iwakura, Y.; Suda, T.; Matsuo, K. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J. Biol. Chem. 2009, 284, 14637–14644. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Kikuta, J.; Ishii, M. Imaging the Bone-Immune Cell Interaction in Bone Destruction. Front. Immunol. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Lorda-Diez, C.I.; Montero, J.A.; Diaz-Mendoza, M.J.; Garcia-Porrero, J.A.; Hurle, J.M. Defining the earliest transcriptional steps of chondrogenic progenitor specification during the formation of the digits in the embryonic limb. PLoS ONE 2011, 6, e24546. [Google Scholar] [CrossRef]
- Wieacker, P.; Wieland, I. Clinical and genetic aspects of craniofrontonasal syndrome: Towards resolving a genetic paradox. Mol. Genet. Metab. 2005, 86, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Kim, J.; Wergedal, J.; Chen, S.T.; Mohan, S. Ephrin B1 regulates bone marrow stromal cell differentiation and bone formation by influencing TAZ transactivation via complex formation with NHERF1. Mol. Cell Biol. 2010, 30, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Arthur, A.; Paton, S.; Hemming, S.; Panagopoulos, R.; Codrington, J.; Walkley, C.R.; Zannettino, A.C.; Gronthos, S. Loss of ephrinB1 in osteogenic progenitor cells impedes endochondral ossification and compromises bone strength integrity during skeletal development. Bone 2016, 93, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Kesavan, C.; Mohan, S.; Qin, X.; Alarcon, C.M.; Wergedal, J.; Xing, W. Transgenic overexpression of ephrin b1 in bone cells promotes bone formation and an anabolic response to mechanical loading in mice. PLoS ONE 2013, 8, e69051. [Google Scholar] [CrossRef]
- Tonna, S.; Poulton, I.J.; Taykar, F.; Ho, P.W.; Tonkin, B.; Crimeen-Irwin, B.; Tatarczuch, L.; McGregor, N.E.; Mackie, E.J.; Martin, T.J.; et al. Chondrocytic ephrin B2 promotes cartilage destruction by osteoclasts in endochondral ossification. Development 2016, 143, 648–657. [Google Scholar] [CrossRef]
- Allan, E.H.; Hausler, K.D.; Wei, T.; Gooi, J.H.; Quinn, J.M.; Crimeen-Irwin, B.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Onyia, J.E.; et al. EphrinB2 regulation by PTH and PTHrP revealed by molecular profiling in differentiating osteoblasts. J. Bone Miner. Res. 2008, 23, 1170–1181. [Google Scholar] [CrossRef]
- Takyar, F.M.; Tonna, S.; Ho, P.W.; Crimeen-Irwin, B.; Baker, E.K.; Martin, T.J.; Sims, N.A. EphrinB2/EphB4 inhibition in the osteoblast lineage modifies the anabolic response to parathyroid hormone. J. Bone Miner. Res. 2013, 28, 912–925. [Google Scholar] [CrossRef]
- Stiffel, V.; Amoui, M.; Sheng, M.H.; Mohan, S.; Lau, K.H. EphA4 receptor is a novel negative regulator of osteoclast activity. J. Bone Miner. Res. 2014, 29, 804–819. [Google Scholar] [CrossRef]
- Arthur, A.; Nguyen, T.M.; Paton, S.; Klisuric, A.; Zannettino, A.C.W.; Gronthos, S. The osteoprogenitor-specific loss of ephrinB1 results in an osteoporotic phenotype affecting the balance between bone formation and resorption. Sci. Rep. 2018, 8, 12756. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zhao, S.L.; Nelson, B.; Kesavan, C.; Qin, X.; Wergedal, J.; Mohan, S.; Xing, W. Targeted disruption of ephrin B1 in cells of myeloid lineage increases osteoclast differentiation and bone resorption in mice. PLoS ONE 2012, 7, e32887. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Baylink, D.; Kesavan, C.; Hu, Y.; Kapoor, S.; Chadwick, R.B.; Mohan, S. Global gene expression analysis in the bones reveals involvement of several novel genes and pathways in mediating an anabolic response of mechanical loading in mice. J. Cell Biochem. 2005, 96, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jong, G.; Lin, L.M.; Shimizu, E. EphB-EphrinB interaction controls odontogenic/osteogenic differentiation with calcium hydroxide. J. Endod. 2013, 39, 1256–1260. [Google Scholar] [CrossRef]
- Goodman, C.S.; Kolodkin, A.L.; Luo, Y.; Püschel, A.W.; Raper, J.A. Unified nomenclature for the semaphorins/collapsins. Semaphorin Nomenclature Committee. Cell 1999, 97, 551–552. [Google Scholar] [CrossRef]
- Epstein, J.A.; Aghajanian, H.; Singh, M.K. Semaphorin signaling in cardiovascular development. Cell Metab. 2015, 21, 163–173. [Google Scholar] [CrossRef]
- Tong, Y.; Chugha, P.; Hota, P.K.; Alviani, R.S.; Li, M.; Tempel, W.; Shen, L.; Park, H.W.; Buck, M. Binding of Rac1, Rnd1, and RhoD to a novel Rho GTPase interaction motif destabilizes dimerization of the plexin-B1 effector domain. J. Biol. Chem. 2007, 282, 37215–37224. [Google Scholar] [CrossRef]
- Toyofuku, T.; Yoshida, J.; Sugimoto, T.; Zhang, H.; Kumanogoh, A.; Hori, M.; Kikutani, H. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat. Neurosci. 2005, 8, 1712–1719. [Google Scholar] [CrossRef]
- Hanna, S.; El-Sibai, M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013, 25, 1955–1961. [Google Scholar] [CrossRef]
- Neufeld, G.; Sabag, A.D.; Rabinovicz, N.; Kessler, O. Semaphorins in angiogenesis and tumor progression. Cold Spring Harb. Perspect. Med. 2012, 2, a006718. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Gemmill, R.M.; Drabkin, H.A. The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. OncoTargets Ther. 2014, 7, 1663–1687. [Google Scholar] [CrossRef]
- Behar, O.; Golden, J.A.; Mashimo, H.; Schoen, F.J.; Fishman, M.C. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature 1996, 383, 525–528. [Google Scholar] [CrossRef]
- Taniguchi, M.; Yuasa, S.; Fujisawa, H.; Naruse, I.; Saga, S.; Mishina, M.; Yagi, T. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 1997, 19, 519–530. [Google Scholar] [CrossRef]
- Gu, C.; Rodriguez, E.R.; Reimert, D.V.; Shu, T.; Fritzsch, B.; Richards, L.J.; Kolodkin, A.L.; Ginty, D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Nakashima, T.; Yoshimura, N.; Okamoto, K.; Tanaka, S.; Takayanagi, H. Autoregulation of Osteocyte Sema3A Orchestrates Estrogen Action and Counteracts Bone Aging. Cell Metab. 2019, 29, 627–637.e625. [Google Scholar] [CrossRef]
- Fukuda, T.; Takeda, S.; Xu, R.; Ochi, H.; Sunamura, S.; Sato, T.; Shibata, S.; Yoshida, Y.; Gu, Z.; Kimura, A.; et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature 2013, 497, 490–493. [Google Scholar] [CrossRef]
- Takegahara, N.; Takamatsu, H.; Toyofuku, T.; Tsujimura, T.; Okuno, T.; Yukawa, K.; Mizui, M.; Yamamoto, M.; Prasad, D.V.; Suzuki, K.; et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol. 2006, 8, 615–622. [Google Scholar] [CrossRef]
- Hughes, A.; Kleine-Albers, J.; Helfrich, M.H.; Ralston, S.H.; Rogers, M.J. A class III semaphorin (Sema3e) inhibits mouse osteoblast migration and decreases osteoclast formation in vitro. Calcif. Tissue Int. 2012, 90, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Delorme, G.; Saltel, F.; Bonnelye, E.; Jurdic, P.; Machuca-Gayet, I. Expression and function of semaphorin 7A in bone cells. Biol. Cell 2005, 97, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Moe, K.; Luukko, K.; Taniguchi, M.; Kettunen, P. Sema3A chemorepellant regulates the timing and patterning of dental nerves during development of incisor tooth germ. Cell Tissue Res. 2014, 357, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Maurin, J.C.; Delorme, G.; Machuca-Gayet, I.; Couble, M.L.; Magloire, H.; Jurdic, P.; Bleicher, F. Odontoblast expression of semaphorin 7A during innervation of human dentin. Matrix Biol. 2005, 24, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Kidd, T.; Bland, K.S.; Goodman, C.S. Slit is the midline repellent for the robo receptor in Drosophila. Cell 1999, 96, 785–794. [Google Scholar] [CrossRef]
- Seeger, M.A.; Beattie, C.E. Attraction versus repulsion: Modular receptors make the difference in axon guidance. Cell 1999, 97, 821–824. [Google Scholar] [CrossRef]
- Brose, K.; Bland, K.S.; Wang, K.H.; Arnott, D.; Henzel, W.; Goodman, C.S.; Tessier-Lavigne, M.; Kidd, T. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 1999, 96, 795–806. [Google Scholar] [CrossRef]
- Dickson, B.J.; Gilestro, G.F. Regulation of commissural axon pathfinding by slit and its Robo receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 651–675. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Duncan, W.C. The SLIT-ROBO pathway: A regulator of cell function with implications for the reproductive system. Reproduction 2010, 139, 697–704. [Google Scholar] [CrossRef]
- Wong, K.; Ren, X.R.; Huang, Y.Z.; Xie, Y.; Liu, G.; Saito, H.; Tang, H.; Wen, L.; Brady-Kalnay, S.M.; Mei, L.; et al. Signal transduction in neuronal migration: Roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell 2001, 107, 209–221. [Google Scholar] [CrossRef]
- Fan, X.; Labrador, J.P.; Hing, H.; Bashaw, G.J. Slit stimulation recruits Dock and Pak to the roundabout receptor and increases Rac activity to regulate axon repulsion at the CNS midline. Neuron 2003, 40, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Bashaw, G.J. Son of sevenless directly links the Robo receptor to rac activation to control axon repulsion at the midline. Neuron 2006, 52, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, E.; Nakamura, H.; Chedotal, A. Repulsive guidance molecule plays multiple roles in neuronal differentiation and axon guidance. J. Neurosci. 2006, 26, 6082–6088. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Tessier-Lavigne, M. Hierarchical organization of guidance receptors: Silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science 2001, 291, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.; Niswander, L. Expression of slit-2 and slit-3 during chick development. Dev. Dyn. 2001, 222, 301–307. [Google Scholar] [CrossRef]
- Kim, B.J.; Lee, Y.S.; Lee, S.Y.; Baek, W.Y.; Choi, Y.J.; Moon, S.A.; Lee, S.H.; Kim, J.E.; Chang, E.J.; Kim, E.Y.; et al. Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J. Clin. Invest. 2018, 128, 1429–1441. [Google Scholar] [CrossRef]
- Li, N.; Inoue, K.; Sun, J.; Niu, Y.; Lalani, S.; Yallowitz, A.; Yang, X.; Zhang, C.; Shen, R.; Zhao, B.; et al. Osteoclasts are not a source of SLIT3. Bone Res. 2020, 8, 11. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.Y.; Lee, S.H.; Koh, J.M.; Kim, B.J. SLIT2 inhibits osteoclastogenesis and bone resorption by suppression of Cdc42 activity. Biochem. Biophys. Res. Commun. 2019, 514, 868–874. [Google Scholar] [CrossRef]
- Sun, H.; Dai, K.; Tang, T.; Zhang, X. Regulation of osteoblast differentiation by slit2 in osteoblastic cells. Cells Tissues Organs 2009, 190, 69–80. [Google Scholar] [CrossRef]
- Xu, R.; Yallowitz, A.; Qin, A.; Wu, Z.; Shin, D.Y.; Kim, J.M.; Debnath, S.; Ji, G.; Bostrom, M.P.; Yang, X.; et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 2018, 24, 823–833. [Google Scholar] [CrossRef]
- Korff, T.; Dandekar, G.; Pfaff, D.; Fuller, T.; Goettsch, W.; Morawietz, H.; Schaffner, F.; Augustin, H.G. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context-dependently with CD31. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Obi, S.; Yamamoto, K.; Shimizu, N.; Kumagaya, S.; Masumura, T.; Sokabe, T.; Asahara, T.; Ando, J. Fluid shear stress induces arterial differentiation of endothelial progenitor cells. J. Appl. Physiol. 2009, 106, 203–211. [Google Scholar] [CrossRef]
- Diercke, K.; Kohl, A.; Lux, C.J.; Erber, R. Strain-dependent up-regulation of ephrin-B2 protein in periodontal ligament fibroblasts contributes to osteogenesis during tooth movement. J. Biol. Chem. 2011, 286, 37651–37664. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Diercke, K.; Zingler, S.; Lux, C.J.; Erber, R. Compression induces Ephrin-A2 in PDL fibroblasts via c-fos. J. Dent. Res. 2015, 94, 464–472. [Google Scholar] [CrossRef]
- Diercke, K.; Sen, S.; Kohl, A.; Lux, C.J.; Erber, R. Compression-dependent up-regulation of ephrin-A2 in PDL fibroblasts attenuates osteogenesis. J. Dent. Res. 2011, 90, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Chen, Y.; Meng, X.; Shi, C.; Li, C.; Chen, Y.; Sun, H. Compressive force regulates ephrinB2 and EphB4 in osteoblasts and osteoclasts contributing to alveolar bone resorption during experimental tooth movement. Korean J. Orthod. 2014, 44, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tao, G.; Guan, Y.; Chen, S.; He, Y.; Li, T.; Zou, S.; Li, Y. The role of ephrinB2-EphB4 signaling on bone remodeling during orthodontic tooth movement. Orthod. Craniofac. Res. 2022. [Google Scholar] [CrossRef]
- Demolli, S.; Doddaballapur, A.; Devraj, K.; Stark, K.; Manavski, Y.; Eckart, A.; Zehendner, C.M.; Lucas, T.; Korff, T.; Hecker, M.; et al. Shear stress-regulated miR-27b controls pericyte recruitment by repressing SEMA6A and SEMA6D. Cardiovasc. Res. 2017, 113, 681–691. [Google Scholar] [CrossRef]
- Sen, S.; Lux, C.J.; Erber, R. A Potential Role of Semaphorin 3A during Orthodontic Tooth Movement. Int. J. Mol. Sci. 2021, 22, 8297. [Google Scholar] [CrossRef]
- Zhang, M.; Ishikawa, S.; Inagawa, T.; Ikemoto, H.; Guo, S.; Sunagawa, M.; Hisamitsu, T. Influence of Mechanical Force on Bone Matrix Proteins in Ovariectomised Mice and Osteoblast-like MC3T3-E1 Cells. In Vivo 2017, 31, 87–95. [Google Scholar] [CrossRef]
- Lin, Y.; Xing, Q.; Qin, W.; de Melo, M.A.S.; Zou, R.; Xu, M.; Zhang, X.; Xu, H.H.K.; Lin, Z. Decreased Expression of Semaphorin3A/Neuropilin-1 Signaling Axis in Apical Periodontitis. Biomed. Res. Int. 2017, 2017, 8724503. [Google Scholar] [CrossRef]
- Song, Y.; Liu, X.; Feng, X.; Gu, Z.; Gu, Y.; Lian, M.; Xiao, J.; Cao, P.; Zheng, K.; Gu, X.; et al. NRP1 Accelerates Odontoblast Differentiation of Dental Pulp Stem Cells Through Classical Wnt/beta-Catenin Signaling. Cell Reprogram. 2017, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Kuchler-Bopp, S.; Bagnard, D.; Van-Der-Heyden, M.; Idoux-Gillet, Y.; Strub, M.; Gegout, H.; Lesot, H.; Benkirane-Jessel, N.; Keller, L. Semaphorin 3A receptor inhibitor as a novel therapeutic to promote innervation of bioengineered teeth. J. Tissue Eng. Regen. Med. 2018, 12, e2151–e2161. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şen, S.; Erber, R. Neuronal Guidance Molecules in Bone Remodeling and Orthodontic Tooth Movement. Int. J. Mol. Sci. 2022, 23, 10077. https://doi.org/10.3390/ijms231710077
Şen S, Erber R. Neuronal Guidance Molecules in Bone Remodeling and Orthodontic Tooth Movement. International Journal of Molecular Sciences. 2022; 23(17):10077. https://doi.org/10.3390/ijms231710077
Chicago/Turabian StyleŞen, Sinan, and Ralf Erber. 2022. "Neuronal Guidance Molecules in Bone Remodeling and Orthodontic Tooth Movement" International Journal of Molecular Sciences 23, no. 17: 10077. https://doi.org/10.3390/ijms231710077
APA StyleŞen, S., & Erber, R. (2022). Neuronal Guidance Molecules in Bone Remodeling and Orthodontic Tooth Movement. International Journal of Molecular Sciences, 23(17), 10077. https://doi.org/10.3390/ijms231710077