
Genome of Laudakia sacra Provides New Insights into High-Altitude Adaptation of Ectotherms

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
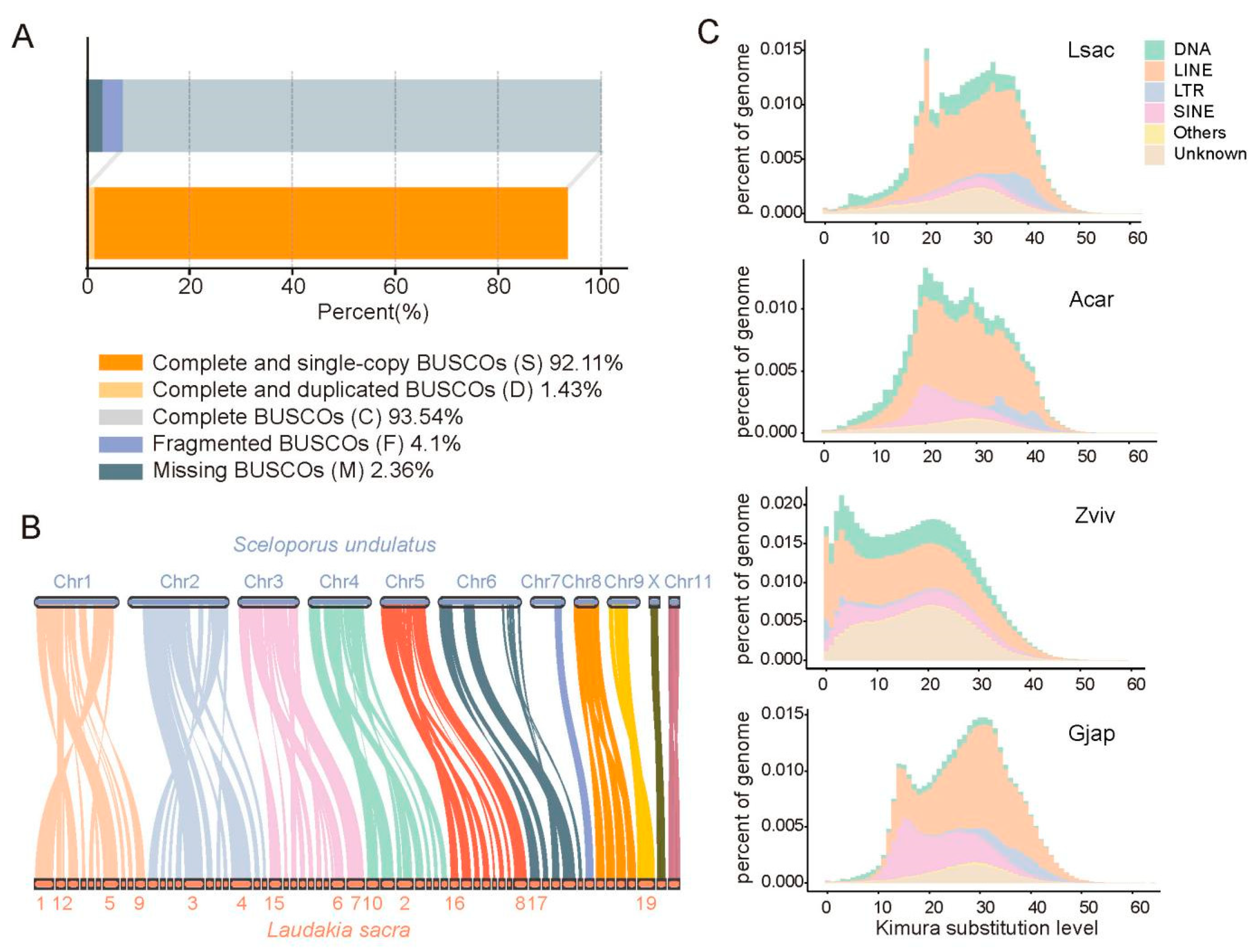
2.1. De Novo Assembly of L. sacra Genome
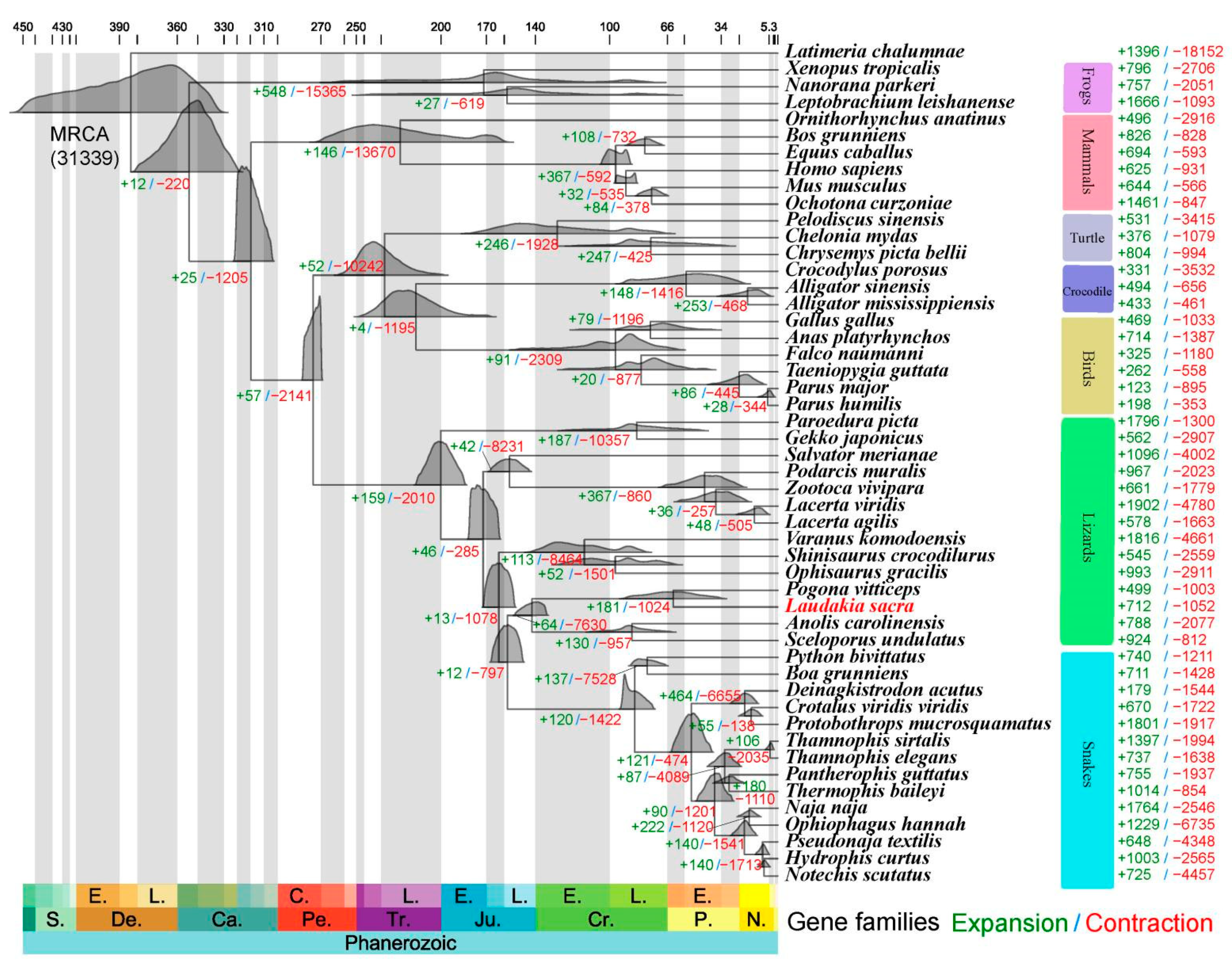
2.2. Phylogenetic Reconstruction
2.3. Positively Selected Genes (PSGs) and Quickly Evolving Genes (QEGs) Analyses
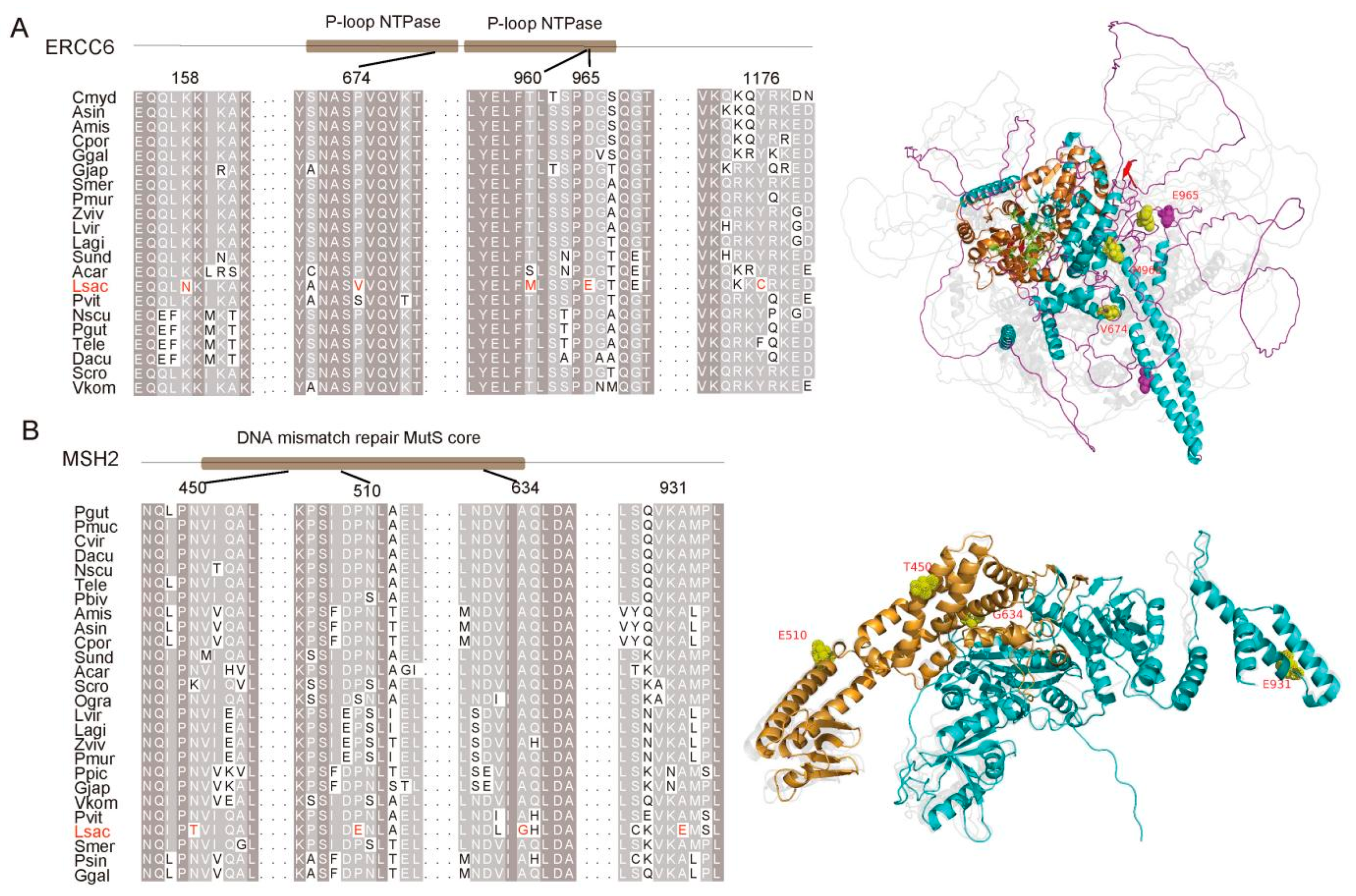
2.4. Potential Genetic Basis of Adaptation to Plateau Environments
3. Discussion
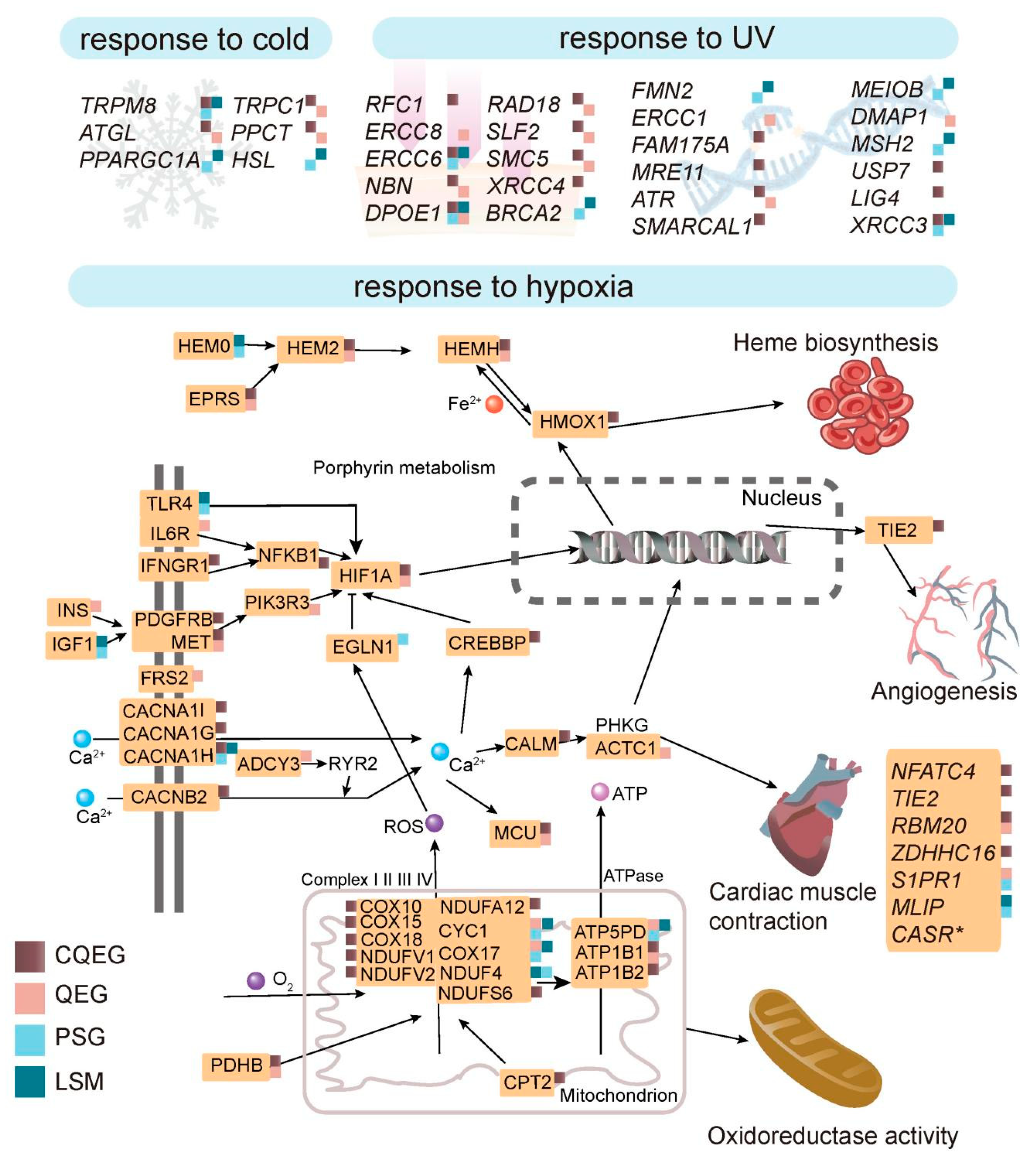
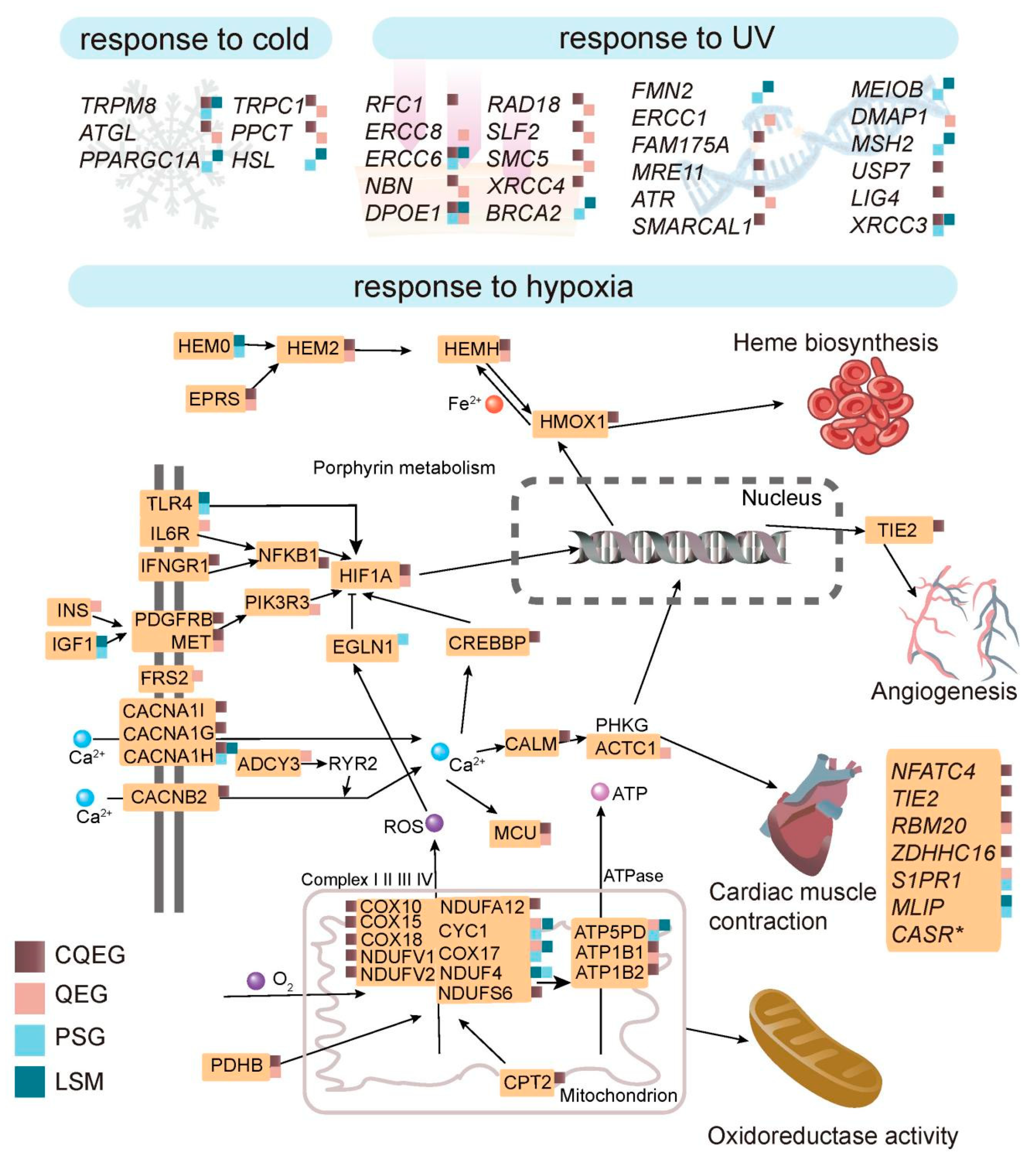
3.1. Adaptation to Hypoxia
3.2. Adaptation to UV Radiation
3.3. Adaptation to Cold
4. Materials and Methods
4.1. Genome Sequencing and Assembly
4.2. Gene Structural and Functional Annotations
4.3. Phylogenetic Tree Construction and Divergence Time Estimation
4.4. Gene Family Expansion and Contraction
4.5. Common Quickly Evolving Genes (CQEGs) in Plateau Animals
4.6. Positively Selected Genes (PSGs) and Quickly Evolving Genes (QEGs) in L. sacra Genome
4.7. Species-Specific Mutations and Alignment Visualization
4.8. Chromosome Synteny with S. undulatus
4.9. Enrichment Analysis and Structural Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michiels, C. Physiological and pathological responses to hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef]
- Sinha, R.P.; Häder, D.-P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.-L.; Cai, Q.; Shen, Y.-Y.; San, A.; Ma, L.; Zhang, Y.; Yi, X.; Chen, Y.; Yang, L.; Huang, Y.; et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 2013, 4, 1858. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-T.; Gao, Y.-D.; Xie, L.; Deng, C.; Shi, P.; Guan, M.-L.; Huang, S.; Ren, J.-L.; Wu, D.-D.; Ding, L.; et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc. Natl. Acad. Sci. USA 2018, 115, 8406–8411. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Han, J.; Tang, X.; Ma, M.; Wang, K.; Zhang, X.; Ren, Q.; Chen, Q.; Qiu, Q. Data from: Comparative transcriptomic analysis revealed adaptation mechanism of Phrynocephalus erythrurus, the highest altitude lizard living in the Qinghai-Tibet Plateau. BMC Evol. Biol. 2015, 15, 101. [Google Scholar] [CrossRef]
- Cai, Q.; Qian, X.; Lang, Y.; Luo, Y.; Xu, J.; Pan, S.; Hui, Y.; Gou, C.; Cai, Y.; Hao, M.; et al. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 2013, 14, R29. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-S.; Li, Y.; Peng, M.-S.; Zhong, L.; Wang, Z.-J.; Li, Q.; Tu, X.-L.; Dong, Y.; Zhu, C.-L.; Wang, L.; et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol. Biol. Evol. 2015, 32, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-B.; Xiong, Z.-J.; Xiang, X.-Y.; Liu, S.-P.; Zhou, W.-W.; Tu, X.-L.; Zhong, L.; Wang, L.; Wu, D.-D.; Zhang, B.-L.; et al. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E1257–E1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-D.; Zhang, B.-L.; Zhou, W.-W.; Li, Y.-X.; Jin, J.-Q.; Shao, Y.; Yang, H.-C.; Liu, Y.-H.; Yan, F.; Chen, H.-M.; et al. Selection and environmental adaptation along a path to speciation in the Tibetan frog Nanorana parkeri. Proc. Natl. Acad. Sci. USA 2018, 115, E5056–E5065. [Google Scholar] [CrossRef]
- Yan, C.; Wu, W.; Dong, W.; Zhu, B.; Chang, J.; Lv, Y.; Yang, S.; Li, J.-T. Temperature acclimation in hot-spring snakes and the convergence of cold response. Innovation 2022, 3, 100295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Phylogenetic relationships and phylogeography of Laudakia sacra and L. wui. Master’s Thesis, Tibet University, Lhasa, China, 2020. [Google Scholar]
- Shi, L.; Huang, S.; Wang, Y. Laudakia sacra. The IUCN Red List of Threatened Species 2019: e.T47751995A47752006. Available online: https://www.iucnredlist.org/species/47751995/47752006 (accessed on 28 July 2022).
- Westfall, A.K.; Telemeco, R.S.; Grizante, M.B.; Waits, D.S.; Clark, A.D.; Simpson, D.Y.; Klabacka, R.L.; Sullivan, A.P.; Perry, G.H.; Sears, M.W.; et al. A chromosome-level genome assembly for the eastern fence lizard (Sceloporus undulatus), a reptile model for physiological and evolutionary ecology. GigaScience 2021, 10, giab066. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.; Stanley, E.L.; Daza, J.D.; Bauer, A.M. A new agamid lizard in mid-Cretaceous amber from northern Myanmar. Cretac. Res. 2021, 124, 104813. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Ramaekers, C.H.; Beucken, T.V.D.; Meng, A.; Kassam, S.; Thoms, J.; Bristow, R.; Wouters, B.G. Hypoxia disrupts the Fanconi anemia pathway and sensitizes cells to chemotherapy through regulation of UBE2T. Radiother. Oncol. 2011, 101, 190–197. [Google Scholar] [CrossRef]
- Scanlon, S.E.; Glazer, P.M. Hypoxic stress facilitates acute activation and chronic downregulation of Fanconi anemia proteins. Mol. Cancer Res. 2014, 12, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Song, H.-P.; Chu, Z.-G.; Zhang, D.-X.; Dang, Y.-M.; Zhang, Q. PI3K–AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by MitoKATP-mediated mitochondrial translocation of pAKT. Cell. Physiol. Biochem. 2018, 49, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.W.; Ribich, S.; Kim, B.W.; Hagen, S.J.; Bianco, A.; Cohen, D.E. Mice lacking Pctp /StarD2 exhibit increased adaptive thermogenesis and enlarged mitochondria in brown adipose tissue. J. Lipid Res. 2009, 50, 2212–2221. [Google Scholar] [CrossRef]
- Rossato, M.; Granzotto, M.; Macchi, V.; Porzionato, A.; Petrelli, L.; Calcagno, A.; Vencato, J.; De Stefani, D.; Silvestrin, V.; Rizzuto, R.; et al. Human white adipocytes express the cold receptor TRPM8 which activation induces UCP1 expression, mitochondrial activation and heat production. Mol. Cell. Endocrinol. 2014, 383, 137–146. [Google Scholar] [CrossRef]
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-induced thermogenesis depends on ATGL-mediated lipolysis in cardiac muscle, but not brown adipose tissue. Cell Metab. 2017, 26, 753–763.e7. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Y.; Zhang, F.; Tang, S.; Zhu, T. Involvement of TRP channels in adipocyte thermogenesis: An update. Front. Cell Dev. Biol. 2021, 9, 1663. [Google Scholar] [CrossRef]
- Broderick, R.; Nieminuszczy, J.; Baddock, H.T.; Deshpande, R.A.; Gileadi, O.; Paull, T.T.; McHugh, P.J.; Niedzwiedz, W. EXD2 promotes homologous recombination by facilitating DNA end resection. Nature 2016, 18, 271–280. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Dianov, G.; Bischoff, C.; Sunesen, M.; Bohr, V.A. Repair of 8-oxoguanine in DNA is deficient in cockayne syndrome group B cells. Nucleic Acids Res. 1999, 27, 1365–1368. [Google Scholar] [CrossRef] [Green Version]
- Keka, I.S.; Mohiuddin; Maede, Y.; Rahman, M.; Sakuma, T.; Honma, M.; Yamamoto, T.; Takeda, S.; Sasanuma, H. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res. 2015, 43, 6359–6372. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.W.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.J.; Demmers, J.A.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.E.; Kim, J.H.; Taylor, M.; Muller, M.T. DNA methyltransferase 1-associated protein (DMAP1) is a co-repressor that stimulates DNA methylation globally and Locally at Sites of Double Strand Break Repair. J. Biol. Chem. 2010, 285, 37630–37640. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; Fragale, A.; Marabitti, V.; Leuzzi, G.; Calcagnile, A.S.; Parlanti, E.; Franchitto, A.; Dogliotti, E.; D’Errico, M. CSA and CSB play a role in the response to DNA breaks. Oncotarget 2018, 9, 11581–11591. [Google Scholar] [CrossRef]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef]
- Popanda, O.; Thielmann, H.W. The function of DNA polymerases in DNA repair synthesis of ultravio-let-irradiated human fibroblasts. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1992, 1129, 155–160. [Google Scholar] [CrossRef]
- Souquet, B.; Abby, E.; Hervé, R.; Finsterbusch, F.; Tourpin, S.; Le Bouffant, R.; Duquenne, C.; Messiaen, S.; Martini, E.; Bernardino-Sgherri, J.; et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013, 9, e1003784. [Google Scholar] [CrossRef]
- Yamada, K.; Ono, M.; Perkins, N.D.; Rocha, S.; Lamond, A.I. Identification and functional characterization of FMN2, a regulator of the cyclin-dependent kinase inhibitor p21. Mol. Cell 2013, 49, 922–933. [Google Scholar] [CrossRef]
- Aravind, L.; Iyer, L.M.; Leipe, D.D.; Koonin, E.V. A novel family of P-loop NTPases with an unusual phyletic distribution and transmembrane segments inserted within the NTPase domain. Genome Biol. 2004, 5, R30. [Google Scholar] [CrossRef]
- Knutson, A.K.; Williams, A.L.; Boisvert, W.A.; Shohet, R.V. HIF in the heart: Development, metabolism, ischemia, and atherosclerosis. J. Clin. Investig. 2021, 131, e137557. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, I.-K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.-C. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, K.; Rey, S.; Zhang, X.; Sebastian, R.; Marti, G.P.; Fox-Talbot, K.; Cardona, A.V.; Du, J.; Tan, Y.S.; Liu, L.; et al. Tie2-dependent knockout of HIF-1 impairs burn wound vascularization and homing of bone marrow-derived angiogenic cells. Cardiovasc. Res. 2012, 93, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Pastore, Y.D.; Divoky, V.; Liu, E.; Mlodnicka, A.E.; Rainey, K.; Ponka, P.; Semenza, G.L.; Schumacher, A.; Prchal, J.T. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J. Biol. Chem. 2006, 281, 25703–25711. [Google Scholar] [CrossRef]
- Abrami, L.; Dallavilla, T.; Sandoz, P.A.; Demir, M.; Kunz, B.; Savoglidis, G.; Hatzimanikatis, V.; van der Goot, F.G. Identification and dynamics of the human ZDHHC16-ZDHHC6 palmitoylation cascade. eLife 2017, 6, e27826. [Google Scholar] [CrossRef] [PubMed]
- Bushdid, P.B.; Osinska, H.; Waclaw, R.R.; Molkentin, J.D.; Yutzey, K.E. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ. Res. 2003, 92, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schäfer, S.; Greaser, M.L.; Radke, M.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Olivey, H.E.; Svensson, E.C. Epicardial–myocardial signaling directing coronary vasculogenesis. Circ. Res. 2010, 106, 818–832. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Schlüter, K.-D. Calcium sensing receptor expression and signalling in cardiovascular physiology and disease. Vasc. Pharmacol. 2018, 107, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.-E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (muscle-enriched A-type lamin-interacting protein) leads to cardiac hyperactivation of Akt/mammalian target of rapamycin (mTOR) and impaired cardiac adaptation. J. Biol. Chem. 2015, 290, 26699–26714. [Google Scholar] [CrossRef]
- Cox, T.C.; Sadlon, T.J.; Schwarz, Q.P.; Matthews, C.S.; Wise, P.D.; Cox, L.L.; Bottomley, S.S.; May, B.K. The major splice variant of human 5-aminolevulinate synthase-2 contributes significantly to erythroid heme biosynthesis. Int. J. Biochem. Cell Biol. 2004, 36, 281–295. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Valnot, I.; Rustin, P.; Taanman, J.-W. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J. Biol. Chem. 2004, 279, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, M.; Sugawara, K.; Tanabe, T.; Yoshida, M. Assembly of human mitochondrial ATP synthase through two separate intermediates, F1-c-ring and b-e-g complex. FEBS Lett. 2015, 589, 2707–2712. [Google Scholar] [CrossRef] [PubMed]
- Cheviron, Z.A.; Bachman, G.C.; Connaty, A.D.; McClelland, G.B.; Storz, J.F. Regulatory changes contribute to the adaptive enhancement of thermogenic capacity in high-altitude deer mice. Proc. Natl. Acad. Sci. USA 2012, 109, 8635–8640. [Google Scholar] [CrossRef]
- Bierer, R.; Nitta, C.H.; Friedman, J.; Codianni, S.; De Frutos, S.; Dominguez-Bautista, J.A.; Howard, T.A.; Resta, T.C.; Bosc, L.V.G. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L872–L880. [Google Scholar] [CrossRef]
- Chen, R.; Yan, J.; Liu, P.; Wang, Z.; Wang, C.; Zhong, W.; Xu, L. The role of nuclear factor of activated T cells in pulmonary arterial hypertension. Cell Cycle 2017, 16, 508–514. [Google Scholar] [CrossRef]
- Jiang, Q.; Geng, X.; Warren, J.; Cosky, E.E.P.; Kaura, S.; Stone, C.; Li, F.; Ding, Y. Hypoxia inducible factor-1α (HIF-1α) mediates NLRP3 inflammasome-dependent-pyroptotic and apoptotic cell death following ischemic stroke. Neuroscience 2020, 448, 126–139. [Google Scholar] [CrossRef]
- Saxena, M.; Eyeretssian, G. NOD-like receptors: Master regulators of inflammation and cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef]
- Wang, H.; Lin, X.; Pu, X. NOD-like receptors mediate inflammatory lung injury during plateau hypoxia exposure. J. Physiol. Anthr. 2020, 39, 32. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, L.; Xiao, R.; Liu, B.; Sun, M.; Liu, F.; Hao, Q.; Lu, Y.; Zhang, J.; Li, J.; et al. Hypoxia-induced mitogenic factor acts as a nonclassical ligand of calcium-sensing receptor, therapeutically exploitable for intermittent hypoxia-induced pulmonary hypertension. Hypertension 2017, 69, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Ghosh, R.K.; Bandyopadhyay, D.; Ghosh, G.C.; Kalra, A.; Lavie, C.J. Significance of pulmonary hypertension in hypertrophic cardiomyopathy. Curr. Probl. Cardiol. 2020, 45, 100398. [Google Scholar] [CrossRef]
- Zhang, F.-L.; Shen, G.-M.; Liu, X.-L.; Wang, F.; Zhao, H.-L.; Yu, J.; Zhang, J.-W. Hypoxic induction of human erythroid-specific δ-aminolevulinate synthase mediated by hypoxia-inducible factor 1. Biochemistry 2011, 50, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.-C.; Tse, A.P.-W.; Law, C.-T.; Xu, I.M.-J.; Lee, D.; Chen, M.; Lai, R.K.-H.; Yuen, V.W.-H.; Cheu, J.W.-S.; Ho, D.W.H.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-H.; Kim, J.-H.; Lee, Y.-H.; Bae, H.C.; Lee, H.-J.; Woo, S.R.; Oh, S.J.; Lee, K.-M.; Yee, C.; Kim, B.W.; et al. Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance. J. Clin. Investig. 2018, 128, 4098–4114. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Walczak-Drzewiecka, A.; Ratajewski, M.; Wagner, W.; Dastych, J. HIF-1α is up-regulated in activated mast cells by a process that involves calcineurin and NFAT. J. Immunol. 2008, 181, 1665–1672. [Google Scholar] [CrossRef]
- Wu, B.; Feng, C.; Zhu, C.; Xu, W.; Yuan, Y.; Hu, M.; Yuan, K.; Li, Y.; Ren, Y.; Zhou, Y.; et al. The genomes of two billfishes provide insights into the evolution of endothermy in teleosts. Mol. Biol. Evol. 2021, 38, 2413–2427. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z. DBG2OLC: Efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Paulini, M.; Twardziok, S.O.; Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018, 19, 189. [Google Scholar] [CrossRef]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Haas, B.J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Han, M.V.; Thomas, G.W.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Polyamines 2014, 1079, 155–170. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Schultz, J. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000, 28, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Alam, M.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-L.; Wu, W.; Tang, C.-Y.; Ren, J.-L.; Jiang, D.; Li, J.-T. Transcriptome analysis reveals olfactory system expression characteristics of aquatic snakes. Front. Genet. 2022, 13, 825974. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, C.; Zhang, Z.-Y.; Lv, Y.; Wang, Z.; Jiang, K.; Li, J.-T. Genome of Laudakia sacra Provides New Insights into High-Altitude Adaptation of Ectotherms. Int. J. Mol. Sci. 2022, 23, 10081. https://doi.org/10.3390/ijms231710081
Yan C, Zhang Z-Y, Lv Y, Wang Z, Jiang K, Li J-T. Genome of Laudakia sacra Provides New Insights into High-Altitude Adaptation of Ectotherms. International Journal of Molecular Sciences. 2022; 23(17):10081. https://doi.org/10.3390/ijms231710081
Chicago/Turabian StyleYan, Chaochao, Zhi-Yi Zhang, Yunyun Lv, Zeng Wang, Ke Jiang, and Jia-Tang Li. 2022. "Genome of Laudakia sacra Provides New Insights into High-Altitude Adaptation of Ectotherms" International Journal of Molecular Sciences 23, no. 17: 10081. https://doi.org/10.3390/ijms231710081
APA StyleYan, C., Zhang, Z.-Y., Lv, Y., Wang, Z., Jiang, K., & Li, J.-T. (2022). Genome of Laudakia sacra Provides New Insights into High-Altitude Adaptation of Ectotherms. International Journal of Molecular Sciences, 23(17), 10081. https://doi.org/10.3390/ijms231710081