
Insights into Allosteric Mechanisms of the Lung-Enriched p53 Mutants V157F and R158L

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
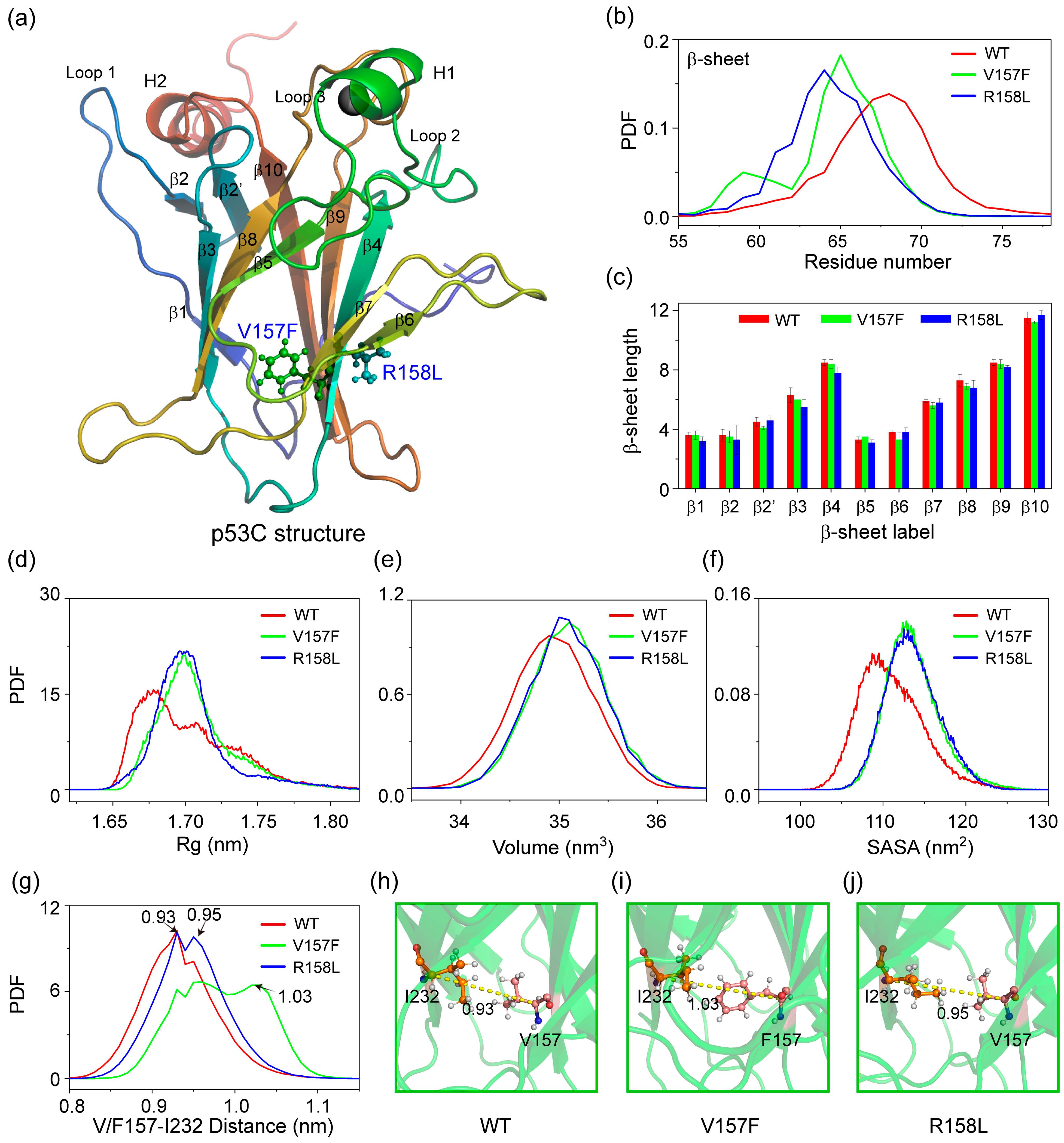
2.1. Both V157F and R158L Mutants Exhibit Structural Features of Aggregation-Prone States
2.2. V157F and R158L Mutation Sites Affect the Conformation Switch of Loop 1 through Long-Range Associations
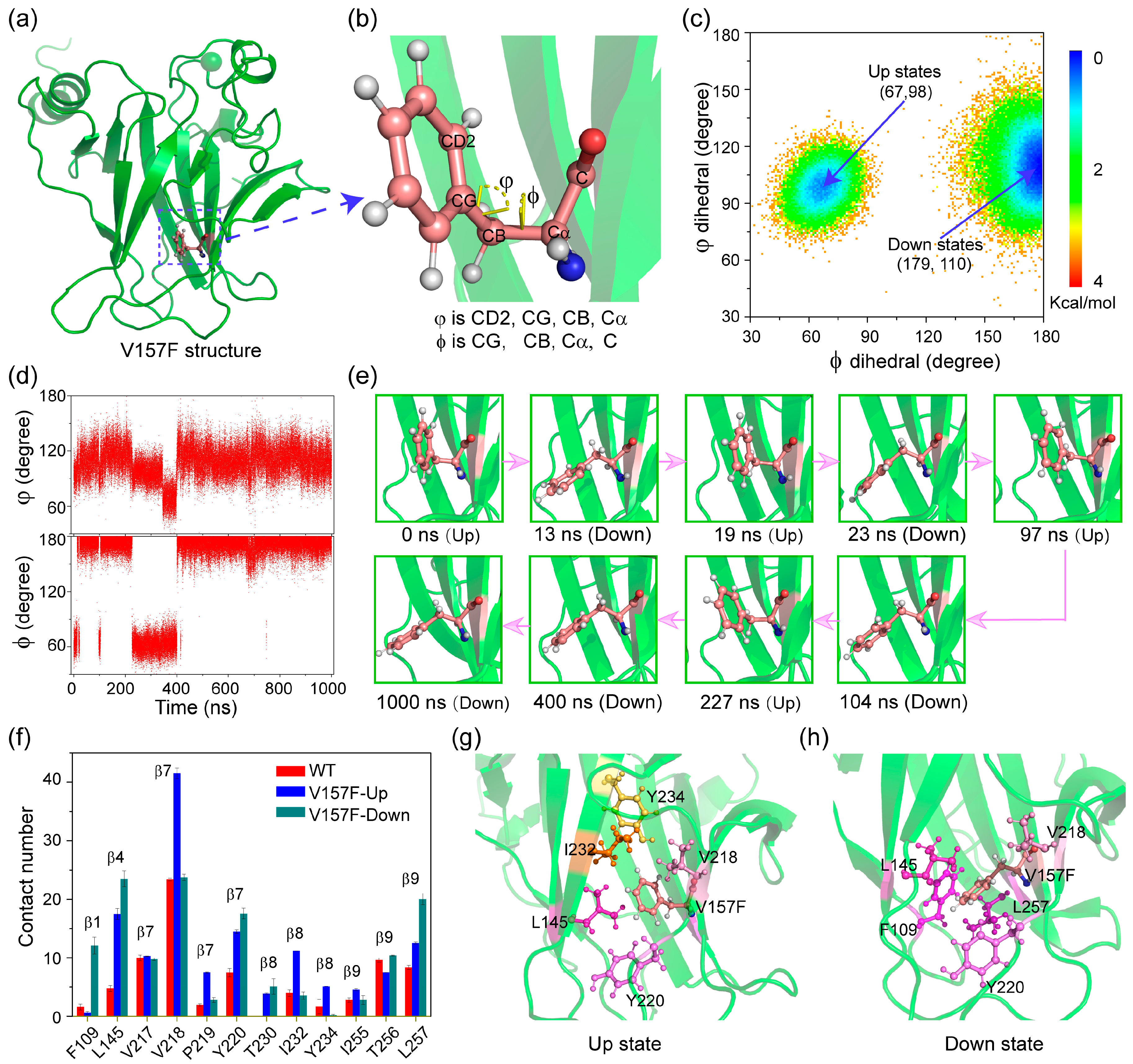
2.3. The Aromatic Ring of Residue F157 Emerges ‘Up’ and ‘Down’ Conformational States in V157F System
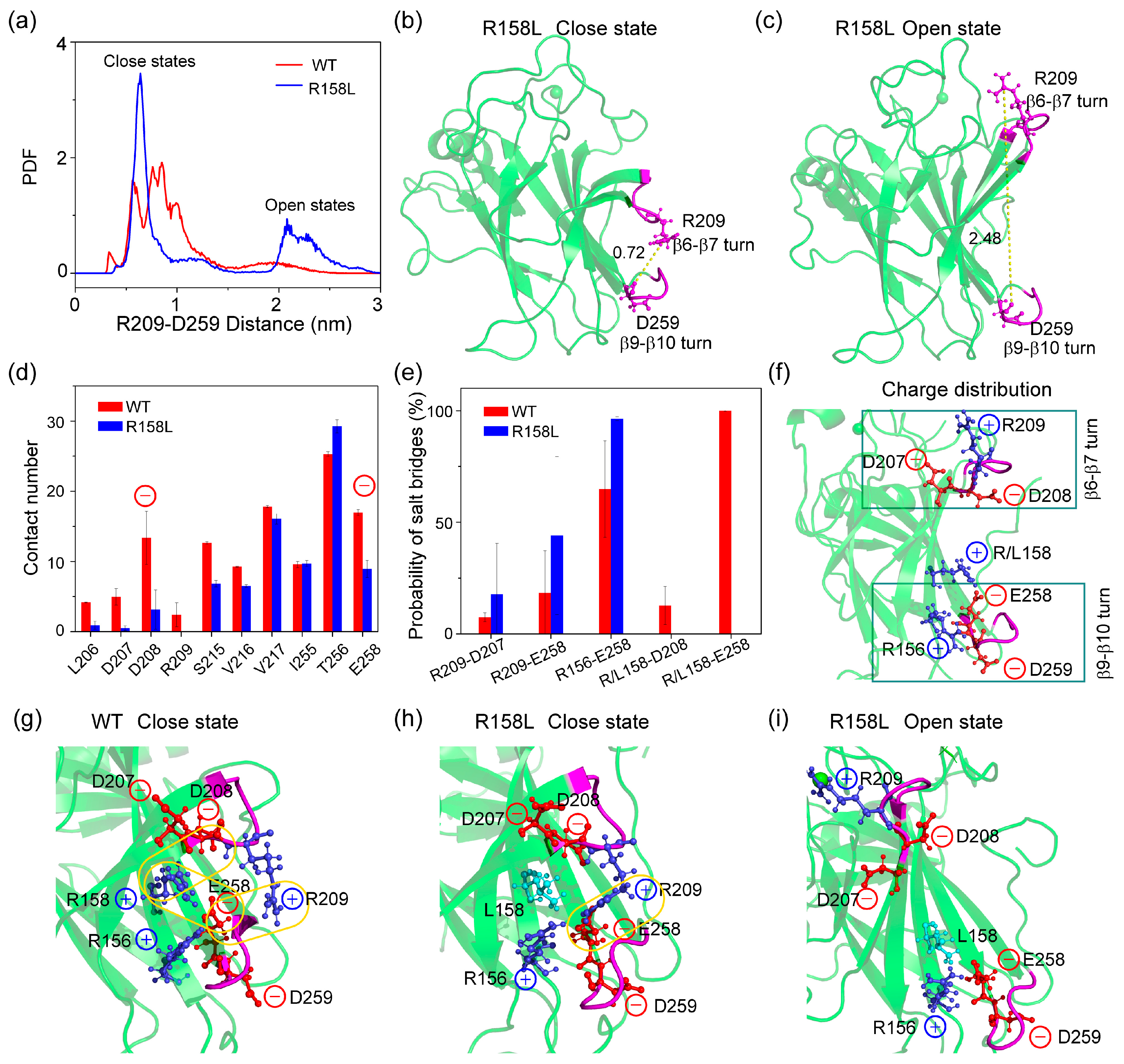
2.4. The β6- β7 Turn Shows ‘Open’ and ‘Close’ States in R158L System Due to the Variation of the Local Salt-Bridge Network
3. Materials and Methods
3.1. WT p53C, V157F and R158L Mutants
3.2. Simulation Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [PubMed]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-function mutant p53: All the roads lead to tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef]
- Friedler, A.; Hansson, L.O.; Veprintsev, D.B.; Freund, S.M.; Rippin, T.M.; Nikolova, P.V.; Proctor, M.R.; Rüdiger, S.; Fersht, A.R. A peptide that binds and stabilizes p53 core domain: Chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. USA 2002, 99, 937–942. [Google Scholar] [CrossRef]
- Canadillas, J.M.; Tidow, H.; Freund, S.M.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Borcherds, W.; Song, T.; Wei, X.; Das, M.; Chen, L.; Daughdrill, G.W.; Chen, J. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc. Natl. Acad. Sci. USA 2019, 116, 8859–8868. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative.e analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Bom, A.P.A.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef]
- Strano, S.; Rossi, M.; Fontemaggi, G.; Munarriz, E.; Soddu, S.; Sacchi, A.; Blandino, G. From p63 to p53 across p73. FEBS Lett. 2001, 490, 163–170. [Google Scholar] [CrossRef]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; Smet, F.D.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Rodin, S.N.; Rodin, A.S. Human lung cancer and p53: The interplay between mutagenesis and selection. Proc. Natl. Acad. Sci. USA 2000, 97, 12244–12249. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Barta, J.A.; McMahon, S.B. Lung-enriched mutations in the p53 tumor suppressor: A paradigm for tissue-specific gain of oncogenic function. Mol. Cancer Res. MCR 2019, 17, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yao, Y.; Wei, G. Unraveling the allosteric mechanism of four cancer-related mutations in the disruption of p53-DNA interaction. J. Phys. Chem. B 2021, 125, 10138–10148. [Google Scholar] [CrossRef] [PubMed]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 amyloids in cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.C.; de Oliveira, G.A.; Cino, E.A.; Soares, I.N.; Rangel, L.P.; Silva, J.L. Aggregation and prion-like properties of misfolded tumor suppressors: Is cancer a prion disease? Cold Spring Harb. Perspect. Biol. 2016, 8, e00054. [Google Scholar] [CrossRef]
- Wang, G.; Fersht, A.R. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA 2017, 114, e2634. [Google Scholar] [CrossRef]
- Pradhan, M.R.; Siau, J.W.; Kannan, S.; Nguyen, M.N.; Ouaray, Z.; Kwoh, C.K.; Lane, D.P.; Ghadessy, F.; Verma, C.S. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019, 47, 1637–1652. [Google Scholar] [CrossRef]
- Li, L.; Li, X.; Tang, Y.; Lao, Z.; Lei, J.; Wei, G. Common cancer mutations R175H and R273H drive the p53 DNA-binding domain towards aggregation-prone conformations. Phys. Chem. Chem. Phys. 2020, 22, 9225–9232. [Google Scholar] [CrossRef]
- Wallentine, B.D.; Wang, Y.; Tretyachenko-Ladokhina, V.; Tan, M.; Senear, D.F.; Luecke, H. Structures of oncogenic, suppressor and rescued p53 core-domain variants: Mechanisms of mutant p53 rescue. Acta Cryst. D Biol. Cryst. 2013, 69, 2146–2156. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Allen, M.D.; Fersht, A.R. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004, 279, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Pedrote, M.M.; de Oliveira, G.A.P.; Felix, A.L.; Mota, M.F.; Marques, M.A.; Soares, I.N.; Iqbal, A.; Norberto, D.R.; Gomes, A.M.O.; Gratton, E.; et al. Aggregation-primed molten globule conformers of the p53 core domain provide potential tools for studying p53C aggregation in cancer. J. Biol. Chem. 2018, 293, 11374–11387. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dunker, A.K.; Powers, J.R.; Clark, S.; Swanson, B.G. Beta-lactoglobulin molten globule induced by high pressure. J. Agric. Food Chem. 2001, 49, 3236–3243. [Google Scholar] [CrossRef]
- Petty, T.J.; Emamzadah, S.; Costantino, L.; Petkova, I.; Stavridi, E.S.; Saven, J.G.; Vauthey, E.; Halazonetis, T.D. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011, 30, 2167–2176. [Google Scholar] [CrossRef]
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop L1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Emamzadah, S.; Tropia, L.; Halazonetis, T.D. Crystal structure of a multidomain human p53 tetramer bound to the natural CDKN1A (p21) p53-response element. Mol. Cancer Res. MCR 2011, 9, 1493–1499. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural survey of zinc-containing proteins and development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Wei, G.; Nussinov, R.; Ma, B. Self-aggregation and coaggregation of the p53 core fragment with its aggregation gatekeeper variant. Phys. Chem. Chem. Phys. 2016, 18, 8098–8107. [Google Scholar] [CrossRef]
- Cino, E.A.; Soares, I.N.; Pedrote, M.M.; de Oliveira, G.A.; Silva, J.L. Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Sci. Rep. 2016, 6, 32535. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Cai, M.; Shen, Y.; Lin, D. Molecular dynamics study on the inhibition mechanisms of ReACp53 peptide for p53-R175H mutant aggregation. Phys. Chem. Chem. Phys. 2021, 23, 23032–23041. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N⋅log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical ve.ersion of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. Lincs: A linear constraint solver for molecular simulations. J. Comput. Chem. 2009, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Day, R.; Bennion, B.J.; Ham, S.; Daggett, V. Increasing temperature accelerates protein unfolding without changing the pathway of unfolding. J. Mol. Biol. 2002, 322, 189–203. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, Y.; Chen, Y.; Lei, J.; Wei, G. Molecular dynamics simulations reveal the mechanism of graphene oxide nanosheet inhibition of Abeta1-42 peptide aggregation. Phys. Chem. Chem. Phys. 2019, 21, 10981–10991. [Google Scholar] [CrossRef]
- Zhan, C.; Lao, Z.; Tang, Y.; Qiao, Q. Natural stereoisomeric flavonoids exhibit different disruptive effects and the mechanism of action on Aβ(42) protofibril. Chem. Commun. 2021, 57, 4267–4270. [Google Scholar] [CrossRef]
- Lei, J.; Qi, R.; Tang, Y.; Wang, W.; Wei, G.; Nussinov, R.; Ma, B. Conformational stability and dynamics of the cancer-associated isoform delta133p53beta are modulated by p53 peptides and p53-specific DNA. FASEB J. 2019, 33, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Till, M.S.; Ullmann, G.M. McVol-A program for calculating protein volumes and identifying cavities by a monte carlo algorithm. J. Mol. Modeling 2010, 16, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Qi, R.; Xie, L.; Xi, W.; Wei, G. Inhibitory effect of hydrophobic fullerenes on the β-sheet-rich oligomers of a hydrophilic GNNQQNY peptide revealed by atomistic simulations. RSC Adv. 2017, 7, 13947–13956. [Google Scholar] [CrossRef]
- Floyd, R.W. Algorithm 97: Shortest path. Commun. ACM 1962, 5, 345. [Google Scholar] [CrossRef]
- Warshall, S. A theorem on boolean matrices. J. ACM 1962, 9, 11–12. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, J.; Li, X.; Cai, M.; Guo, T.; Lin, D.; Deng, X.; Li, Y. Insights into Allosteric Mechanisms of the Lung-Enriched p53 Mutants V157F and R158L. Int. J. Mol. Sci. 2022, 23, 10100. https://doi.org/10.3390/ijms231710100
Lei J, Li X, Cai M, Guo T, Lin D, Deng X, Li Y. Insights into Allosteric Mechanisms of the Lung-Enriched p53 Mutants V157F and R158L. International Journal of Molecular Sciences. 2022; 23(17):10100. https://doi.org/10.3390/ijms231710100
Chicago/Turabian StyleLei, Jiangtao, Xuanyao Li, Mengqiang Cai, Tianjing Guo, Dongdong Lin, Xiaohua Deng, and Yin Li. 2022. "Insights into Allosteric Mechanisms of the Lung-Enriched p53 Mutants V157F and R158L" International Journal of Molecular Sciences 23, no. 17: 10100. https://doi.org/10.3390/ijms231710100
APA StyleLei, J., Li, X., Cai, M., Guo, T., Lin, D., Deng, X., & Li, Y. (2022). Insights into Allosteric Mechanisms of the Lung-Enriched p53 Mutants V157F and R158L. International Journal of Molecular Sciences, 23(17), 10100. https://doi.org/10.3390/ijms231710100