
Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
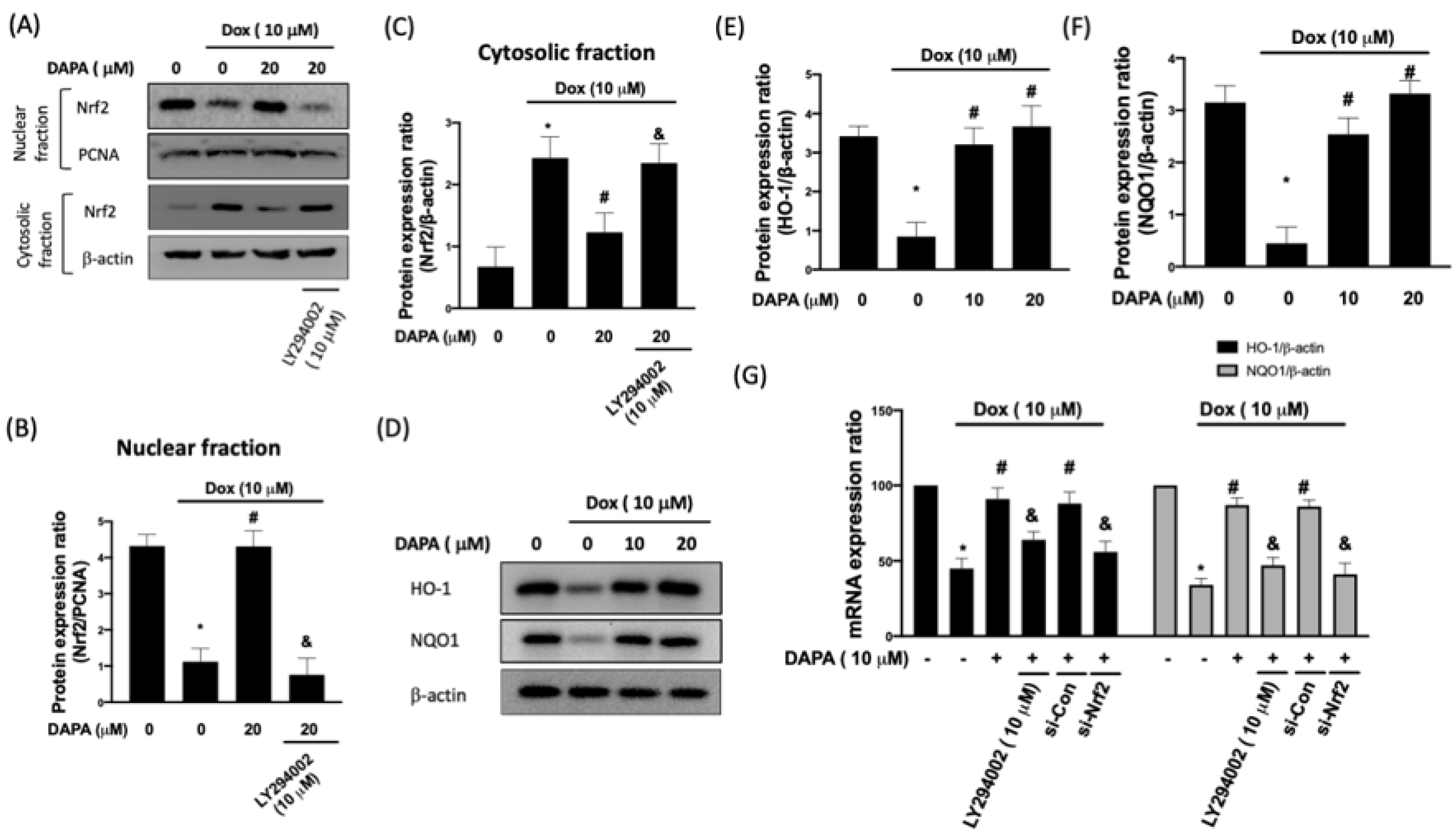
2.1. DAPA Reverses the Dox-Induced Repression of HO-1 and NQO1 via PI3K/AKT/Nrf2 Axis in H9c2 Cells
2.2. DAPA Restores the DOX-Induced Downregulation of Antioxidant Capacity and Mitochondrial Dysfunction in H9c2 Cells
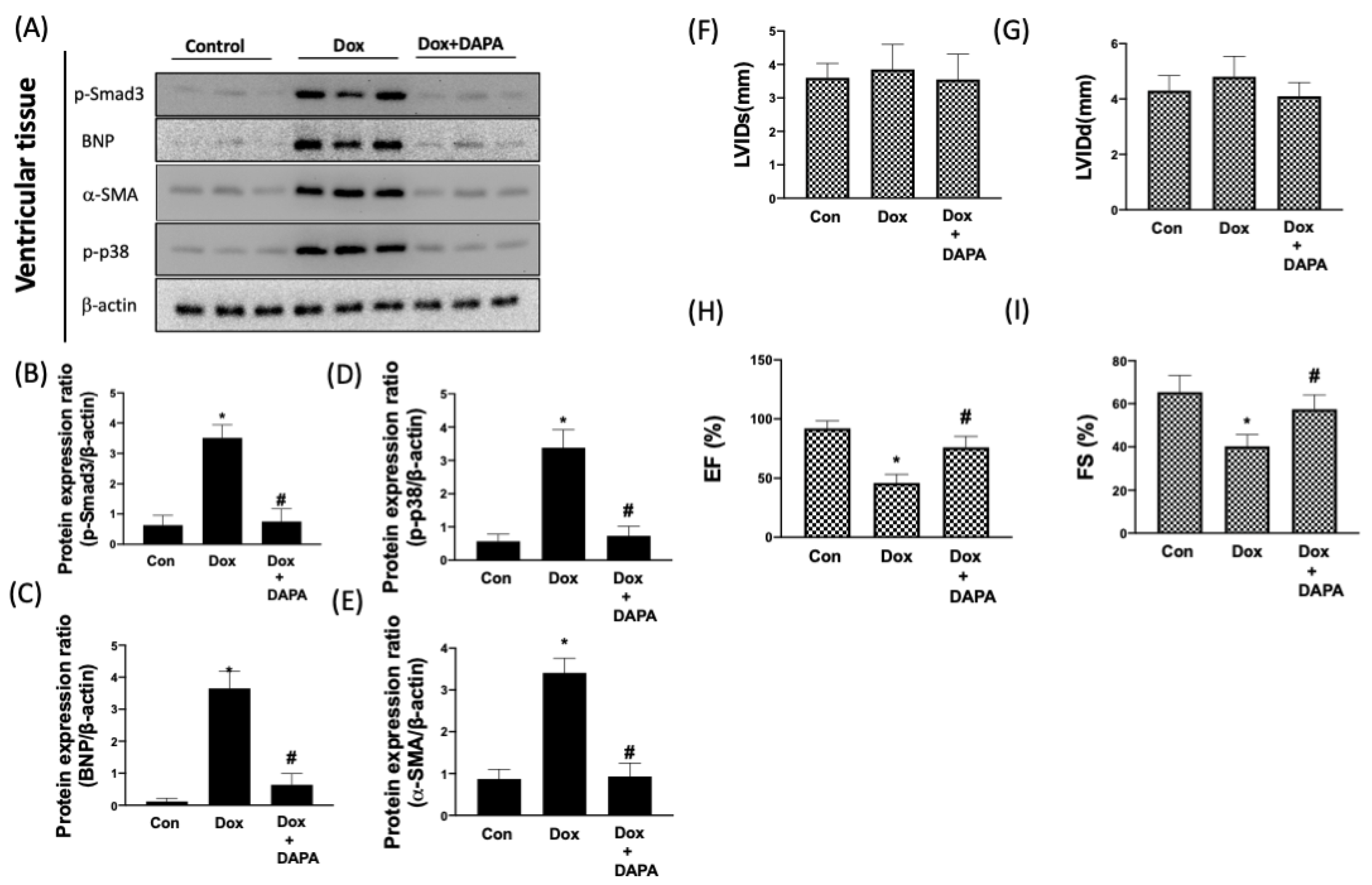
2.3. DAPA Suppresses the DOX-Elicited Cardiac Remodeling
2.4. DAPA Ameliorates the DOX-Stimulated Cardiac Inflammation in H9c2 Cells
2.5. Cardioprotective Effect of DAPA in Dox-Injected Rats
3. Discussion
4. Materials and Methods
4.1. Cell Culture and In Vivo Model
4.2. Reagents
4.3. Cell Viability
4.4. Primary Culture of Cardiomyocytes
4.5. Western Blotting Assay
4.6. RNA Isolation and Analysis
4.7. Investigation of Mitochondrial Membrane Potential
4.8. Cytoplasmic and Nuclear Protein Extraction and NF-kB Activity
4.9. IL-8 Concentration and SOD Activity
4.10. Investigation of ROS Production
4.11. Transfection with Small Interfering RNA
4.12. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, G.C.; Jordan, J.H.; D’Agostino, R.B., Jr.; Vasu, S.; Hamilton, C.A.; Hundley, W.G. Progressive 3-Month Increase in LV Myocardial ECV After Anthracycline-Based Chemotherapy. JACC Cardiovasc. Imaging 2017, 10, 708–709. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Chang, W.T.; Shih, J.Y.; Lin, Y.W.; Chen, Z.C.; Kan, W.C.; Lin, T.H.; Hong, C.S. Dapagliflozin protects against doxorubicin-induced cardiotoxicity by restoring STAT3. Arch. Toxicol. 2022, 96, 2021–2032. [Google Scholar] [CrossRef]
- Chang, W.T.; Lin, Y.W.; Ho, C.H.; Chen, Z.C.; Liu, P.Y.; Shih, J.Y. Dapagliflozin suppresses ER stress and protects doxorubicin-induced cardiotoxicity in breast cancer patients. Arch. Toxicol. 2021, 95, 659–671. [Google Scholar] [CrossRef]
- Kitamura, Y.; Koide, M.; Akakabe, Y.; Matsuo, K.; Shimoda, Y.; Soma, Y.; Ogata, T.; Ueyama, T.; Matoba, S.; Yamada, H.; et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J. Biol. Chem. 2014, 289, 2788–2800. [Google Scholar] [CrossRef]
- Fan, G.C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef]
- El-Sayed, N.; Mostafa, Y.M.; AboGresha, N.M.; Ahmed, A.A.M.; Mahmoud, I.Z.; El-Sayed, N.M. Dapagliflozin attenuates diabetic cardiomyopathy through erythropoietin up-regulation of AKT/JAK/MAPK pathways in streptozotocin-induced diabetic rats. Chem. Biol. Interact. 2021, 347, 109617. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Niu, T.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Li, H.; Janicki, J.S.; Wang, X.L.; et al. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid. Med. Cell Longev. 2014, 2014, 748524. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arter. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef]
- Dreger, H.; Westphal, K.; Weller, A.; Baumann, G.; Stangl, V.; Meiners, S.; Stangl, K. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovasc. Res. 2009, 83, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. 2015, 4, e002423. [Google Scholar] [CrossRef]
- Guo, R.M.; Xu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-κB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef]
- Damås, J.K.; Gullestad, L.; Ueland, T.; Solum, N.O.; Simonsen, S.; Frøland, S.S.; Aukrust, P. CXC-chemokines, a new group of cytokines in congestive heart failure--possible role of platelets and monocytes. Cardiovasc. Res. 2000, 45, 428–436. [Google Scholar] [CrossRef]
- Yang, H.L.; Hsieh, P.L.; Hung, C.H.; Cheng, H.C.; Chou, W.C.; Chu, P.M.; Chang, Y.C.; Tsai, K.L. Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-Caused Cardiac Dysfunction Through Inhibition of Cardiac Fibrosis and Inflammation. Cancers 2020, 12, 1102. [Google Scholar] [CrossRef]
- Kunsch, C.; Lang, R.K.; Rosen, C.A.; Shannon, M.F. Synergistic transcriptional activation of the IL-8 gene by NF-kappa B p65 (RelA) and NF-IL-6. J. Immunol. 1994, 153, 153–164. [Google Scholar] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Soto-Pantoja, D.R.; Bi, J.; Hundley, W.G.; Widiapradja, A.; Manteufel, E.J.; Bradshaw, T.W.; Meléndez, G.C. Doxorubicin-Induced Myocardial Fibrosis Involves the Neurokinin-1 Receptor and Direct Effects on Cardiac Fibroblasts. Heart Lung Circ. 2019, 28, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes. Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Arab, H.H.; Safar, M.M.; Shahin, N.N. Targeting ROS-Dependent AKT/GSK-3β/NF-κB and DJ-1/Nrf2 Pathways by Dapagliflozin Attenuates Neuronal Injury and Motor Dysfunction in Rotenone-Induced Parkinson’s Disease Rat Model. ACS Chem. Neurosci. 2021, 12, 689–703. [Google Scholar] [CrossRef]
- Yang, L.; Liu, D.; Yan, H.; Chen, K. Dapagliflozin attenuates cholesterol overloading-induced injury in mice hepatocytes with type 2 diabetes mellitus (T2DM) via eliminating oxidative damages. Cell Cycle 2022, 21, 641–654. [Google Scholar] [CrossRef]
- Hazem, R.M.; Ibrahim, A.Z.; Ali, D.A.; Moustafa, Y.M. Dapagliflozin improves steatohepatitis in diabetic rats via inhibition of oxidative stress and inflammation. Int. Immunopharmacol. 2022, 104, 108503. [Google Scholar] [CrossRef]
- Abd El-Fattah, E.E.; Saber, S.; Mourad, A.A.E.; El-Ahwany, E.; Amin, N.A.; Cavalu, S.; Yahya, G.; Saad, A.S.; Alsharidah, M.; Shata, A.; et al. The dynamic interplay between AMPK/NFκB signaling and NLRP3 is a new therapeutic target in inflammation: Emerging role of dapagliflozin in overcoming lipopolysaccharide-mediated lung injury. Biomed. Pharm. 2022, 147, 112628. [Google Scholar] [CrossRef]
- Sardão, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Morphological alterations induced by doxorubicin on H9c2 myoblasts: Nuclear, mitochondrial, and cytoskeletal targets. Cell Biol. Toxicol. 2009, 25, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Sardão, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharm. 2009, 64, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Starinets, V.S.; Belosludtsev, M.N.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Chronic treatment with dapagliflozin protects against mitochondrial dysfunction in the liver of C57BL/6NCrl mice with high-fat diet/streptozotocin-induced diabetes mellitus. Mitochondrion 2021, 59, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Hsieh, P.L.; Chou, W.C.; Cheng, H.C.; Huang, Y.T.; Chan, S.H. Dapagliflozin attenuates hypoxia/reoxygenation-caused cardiac dysfunction and oxidative damage through modulation of AMPK. Cell Biosci. 2021, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.K.; Ahmed, A.; Singla, R.; Yan, B. Embryonic stem cells improve cardiac function in Doxorubicin-induced cardiomyopathy mediated through multiple mechanisms. Cell Transpl. 2012, 21, 1919–1930. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef]
- Tanaka, R.; Umemura, M.; Narikawa, M.; Hikichi, M.; Osaw, K.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Tamura, K.; Ishikawa, Y. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020, 7, 588–603. [Google Scholar] [CrossRef]
- Nousiainen, T.; Vanninen, E.; Jantunen, E.; Puustinen, J.; Remes, J.; Rantala, A.; Vuolteenaho, O.; Hartikainen, J. Natriuretic peptides during the development of doxorubicin-induced left ventricular diastolic dysfunction. J. Intern. Med. 2002, 251, 228–234. [Google Scholar] [CrossRef]
- Butt, J.H.; Adamson, C.; Docherty, K.F.; de Boer, R.A.; Petrie, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; Maria Langkilde, A.; Lindholm, D.; Martinez, F.A.; et al. Efficacy and Safety of Dapagliflozin in Heart Failure with Reduced Ejection Fraction according to N-Terminal Pro-B-Type Natriuretic Peptide: Insights from the DAPA-HF Trial. Circ. Heart Fail. 2021, 14, e008837. [Google Scholar] [CrossRef]
- Jarai, R.; Kaun, C.; Weiss, T.W.; Speidl, W.S.; Rychli, K.; Maurer, G.; Huber, K.; Wojta, J. Human cardiac fibroblasts express B-type natriuretic peptide: Fluvastatin ameliorates its up-regulation by interleukin-1alpha, tumour necrosis factor-alpha and transforming growth factor-beta. J. Cell Mol. Med. 2009, 13, 4415–4421. [Google Scholar] [CrossRef] [Green Version]
- Nymo, S.H.; Hulthe, J.; Ueland, T.; McMurray, J.; Wikstrand, J.; Askevold, E.T.; Yndestad, A.; Gullestad, L.; Aukrust, P. Inflammatory cytokines in chronic heart failure: Interleukin-8 is associated with adverse outcome. Results from CORONA. Eur. J. Heart Fail. 2014, 16, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, Z.; Sun, Y.; Liu, X.; Ma, W.; Ding, Y.; Hong, J.; Qian, L.; Xu, D. Dapagliflozin Improves Cardiac Function, Remodeling, Myocardial Apoptosis, and Inflammatory Cytokines in Mice with Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, P.-L.; Chu, P.-M.; Cheng, H.-C.; Huang, Y.-T.; Chou, W.-C.; Tsai, K.-L.; Chan, S.-H. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. Int. J. Mol. Sci. 2022, 23, 10146. https://doi.org/10.3390/ijms231710146
Hsieh P-L, Chu P-M, Cheng H-C, Huang Y-T, Chou W-C, Tsai K-L, Chan S-H. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. International Journal of Molecular Sciences. 2022; 23(17):10146. https://doi.org/10.3390/ijms231710146
Chicago/Turabian StyleHsieh, Pei-Ling, Pei-Ming Chu, Hui-Ching Cheng, Yu-Ting Huang, Wan-Ching Chou, Kun-Ling Tsai, and Shih-Hung Chan. 2022. "Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation" International Journal of Molecular Sciences 23, no. 17: 10146. https://doi.org/10.3390/ijms231710146
APA StyleHsieh, P.-L., Chu, P.-M., Cheng, H.-C., Huang, Y.-T., Chou, W.-C., Tsai, K.-L., & Chan, S.-H. (2022). Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. International Journal of Molecular Sciences, 23(17), 10146. https://doi.org/10.3390/ijms231710146