
Chrysin-Induced G Protein-Coupled Estrogen Receptor Activation Suppresses Pancreatic Cancer

 ,
, 
Abstract
:1. Introduction
2. Results
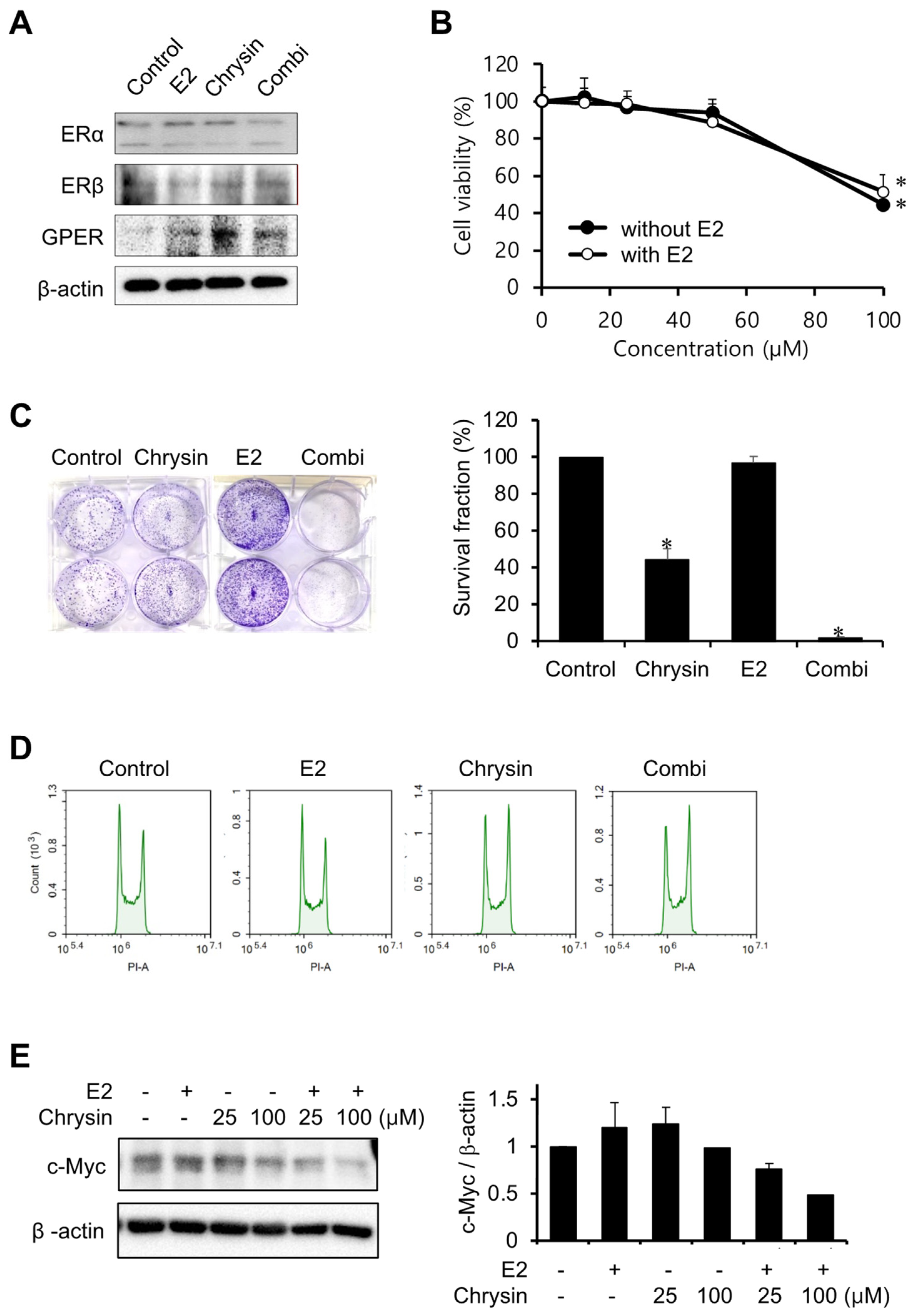
2.1. Cytotoxic Effect of Chrysin in MIA PaCa-2 Cells
2.2. Effect of E2 on Chrysin-Related Inhibition of MIA PaCa-2 Cell Proliferation
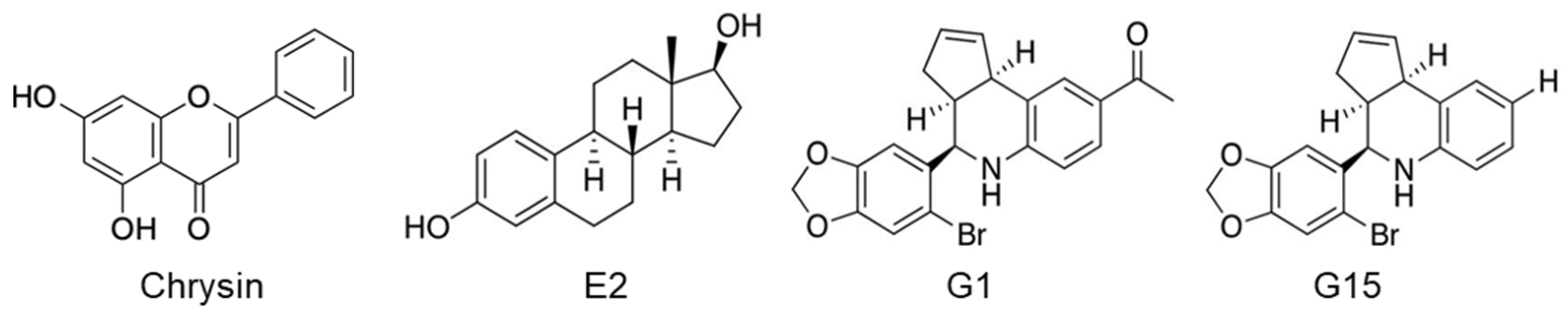
2.3. Inhibition of Cell Proliferation Using a GPER Agonist
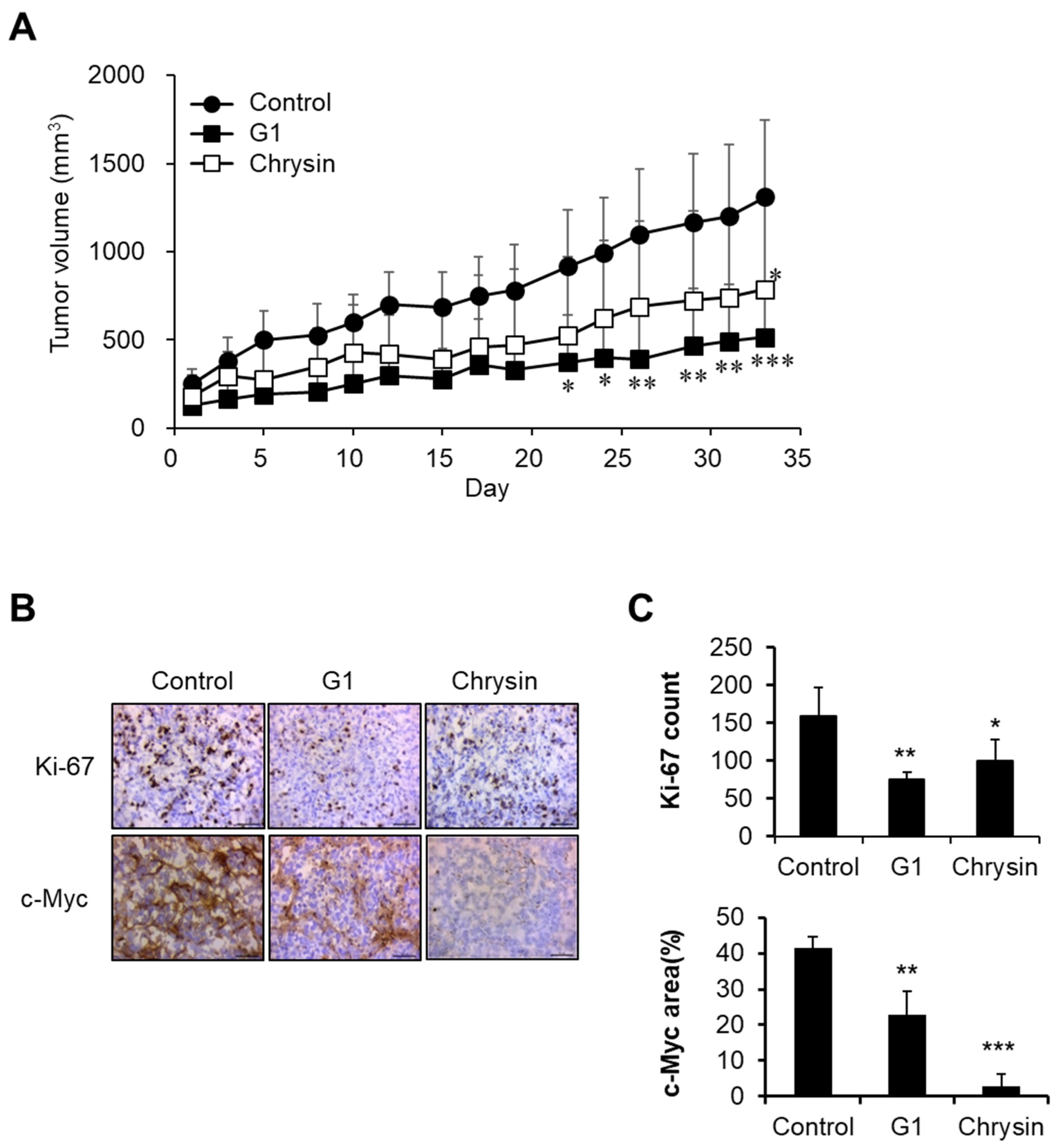
2.4. Chrysin-Related Tumor Growth Delay in a MIA PaCa-2-Cell-Derived Xenograft Model
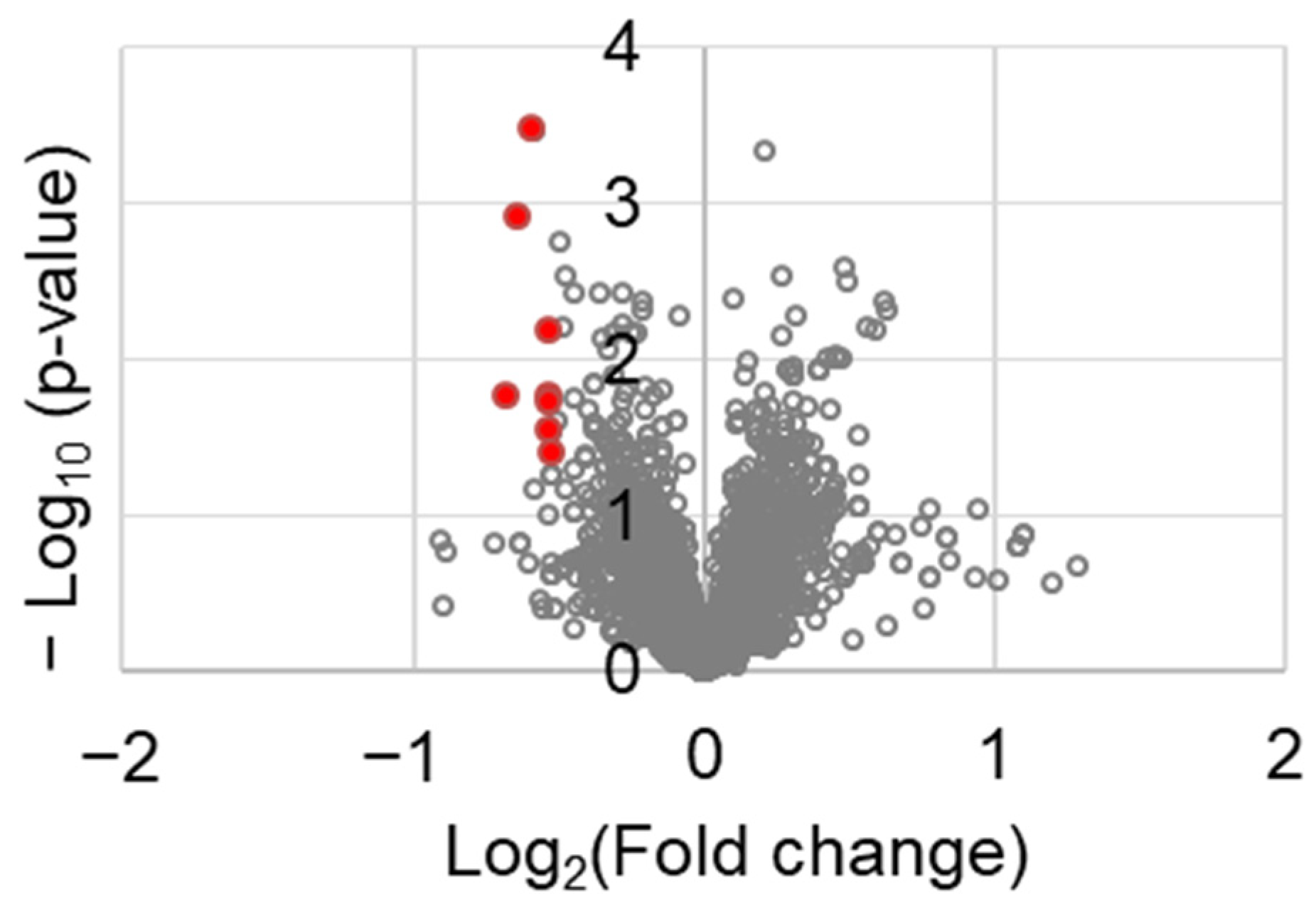
2.5. Chrysin-Associated Molecular Factors in the Inhibition of Pancreatic Cancer Progression
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Clonogenic Assay
4.5. Western Blot Analysis
4.6. Cell Cycle Analysis
4.7. Tumor Growth
4.8. Immunohistochemistry
4.9. Proteomics Data Acquisition
4.10. Overall Survival (OS) Analysis of Differentially Expressed Genes (DEGs)
4.11. Protein-Protein Interaction
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer. J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-X.; Zhao, C.-F.; Chen, W.-B.; Liu, Q.-C.; Li, Q.-W.; Lin, Y.-Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Won, Y.-J.; Lee, J.J.; Jung, K.-W.; Kong, H.-J.; Im, J.-S.; Seo, H.G. Cancer Statistics in Korea: Incidence, mortality, survival, and prevalence in 2018. Cancer. Res. Treat. 2021, 53, 301–315. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer. J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Reni, M.; Cordio, S.; Milandri, C.; Passoni, P.; Bonetto, E.; Oliani, C.; Luppi, G.; Nicoletti, R.; Galli, L.; Bordonaro, R.; et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: A randomised controlled multicentre phase III trial. Lancet Oncol. 2005, 6, 369–376. [Google Scholar] [CrossRef]
- Li, J.; Wientjes, M.G.; Au, J.L.S. Pancreatic cancer: Pathobiology, treatment options, and drug delivery. AAPS J. 2010, 12, 223–232. [Google Scholar] [CrossRef]
- Cipolletti, M.; Solar Fernandez, V.; Montalesi, E.; Marino, M.; Fiocchetti, M. Beyond the antioxidant activity of dietary polyphenols in cancer: The modulation of estrogen receptors (ERs) Signaling. Int. J. Mol. Sci. 2018, 19, 2624. [Google Scholar] [CrossRef]
- Sellitto, A.; D’Agostino, Y.; Alexandrova, E.; Lamberti, J.; Pecoraro, G.; Memoli, D.; Rocco, D.; Coviello, E.; Giurato, G.; Nassa, G.; et al. Insights into the role of estrogen receptor β in triple-negative breast cancer. Cancers 2020, 12, 1477. [Google Scholar] [CrossRef]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Benz, C.; Hollander, C.; Miller, B. Endocrine-responsive pancreatic carcinoma: Steroid binding and cytotoxicity studies in human tumor cell lines. Cancer Res. 1986, 46, 2276–2281. [Google Scholar]
- Guo, J.-M.; Xiao, B.-X.; Dai, D.-J.; Liu, Q.; Ma, H.-H. Effects of daidzein on estrogen-receptor-positive and negative pancreatic cancer cells in vitro. World J. Gastroenterol. 2004, 10, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Horn-Ross, P.L.; Rull, R.P.; Neuhausen, S.L.; Anton-Culver, H.; Ursin, G.; Henderson, K.D.; Bernstein, L. Reproductive factors, exogenous hormones, and pancreatic cancer risk in the CTS. Am. J. Epidemiol. 2013, 178, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Sadr-Azodi, O.; Konings, P.; Brusselaers, N. Menopausal hormone therapy and pancreatic cancer risk in women: A population-based matched cohort study. United Eur. Gastroenterol. J. 2017, 5, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G protein-coupled estrogen receptor: A potential therapeutic target in cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Gupta, V.K.; Banerjee, S.; Saluja, A.K. Learning rrom gender disparity: Role of estrogen receptor activation in coping with pancreatic cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 862–863. [Google Scholar] [CrossRef]
- Chaturantabut, S.; Shwartz, A.; Evason, K.J.; Cox, A.G.; Labella, K.; Schepers, A.G.; Yang, S.; Acuña, M.; Houvras, Y.; Mancio-Silva, L.; et al. Estrogen activation of G-protein-coupled estrogen receptor 1 regulates phosphoinositide 3-kinase and mTOR signaling to promote liver growth in Zebrafish and proliferation of human hepatocytes. Gastroenterology 2019, 156, 1788–1804.e13. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in breast cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; Liu, H.; Du, J.; Wang, H. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Choo, J.H.; Lee, S.H. The Effect of chrysin on the transcriptional activity of vitamin D receptor in human keratinocytes. J. Soc. Cosmet. Sci. Korea 2013, 39, 75–81. [Google Scholar]
- Han, D.-H.; Denison, M.S.; Tachibana, H.; Yamada, K. Relationship between estrogen receptor-binding and estrogenic activities of environmental estrogens and suppression by flavonoids. Biosci. Biotechnol. Biochem. 2002, 66, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Jung, J. Emerging utilization of chrysin using nanoscale modification. J. Nanomater. 2016, 2016, 2894089. [Google Scholar] [CrossRef]
- Lim, H.K.; Kim, K.M.; Jeong, S.-Y.; Choi, E.K.; Jung, J. Chrysin increases the therapeutic efficacy of docetaxel and mitigates docetaxel-induced edema. Integr. Cancer Ther. 2017, 16, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Lim, H.K.; Shim, S.H.; Jung, J. Improved chemotherapeutic efficacy of injectable chrysin encapsulated by copolymer nanoparticles. Int. J. Nanomed. 2017, 12, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Jung, J. Upregulation of G protein-coupled estrogen receptor by chrysin-nanoparticles inhibits tumor proliferation and metastasis in triple negative breast cancer xenograft model. Front. Endocrinol. 2020, 11, 560605. [Google Scholar] [CrossRef] [PubMed]
- Moertl, S.; Payer, S.; Kell, R.; Winkler, K.; Anastasov, N.; Atkinson, M.J. Comparison of radiosensitization by HDAC inhibitors CUDC-101 and SAHA in pancreatic cancer cells. Int. J. Mol. Sci. 2019, 20, 3259. [Google Scholar] [CrossRef]
- Konduri, S.; Schwarz, R.E. Estrogen receptor implications in human pancreatic cancer. In Female Sex Hormones and Cancers; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; pp. 187–207. [Google Scholar]
- Satake, M.; Sawai, H.; Go, V.L.W.; Satake, K.; Reber, H.A.; Hines, O.J.; Eibl, G. Estrogen receptors in pancreatic tumors. Pancreas 2006, 33, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Kostakis, I.D.; Nikolidakis, L.; Kostakis, A.; Kouraklis, G. Nuclear receptors in pancreatic tumor cells. Anticancer Res. 2014, 34, 6897–6911. [Google Scholar]
- Konduri, S.; Schwarz, R.E. Estrogen receptor β/α ratio predicts response of pancreatic cancer cells to estrogens and phytoestrogens. J. Surg. Res. 2007, 140, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kang, M.-J.; Lee, S.H.; Son, J.H.; Lee, J.E.; Paik, W.H.; Ryu, J.K.; Kim, Y.-T. Fisetin enhances the cytotoxicity of gemcitabine by down-regulating ERK-MYC in MiaPaca-2 human pancreatic cancer cells. Anticancer Res. 2018, 38, 3527–3533. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Qu, C.; Chen, L.; Geng, Y.; Cheng, C.; Yu, S.; Wang, D.; Yang, L.; Meng, Z.; et al. Kaempferol induces ROS-dependent apoptosis in pancreatic cancer cells via TGM2-mediated Akt/mTOR signaling. BMC Cancer 2021, 21, 396. [Google Scholar] [CrossRef]
- Wang, W.; VanAlstyne, P.C.; Irons, K.A.; Chen, S.; Stewart, J.W.; Birt, D.F. Individual and interactive effects of apigenin analogs on G2/M cell-cycle arrest in human colon carcinoma cell lines. Nutr. Cancer 2004, 48, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Von Brandenstein, M.G.; Abety, A.N.; Depping, R.; Roth, T.; Koehler, M.; Dienes, H.-P.; Fries, J.W.U. A p38–p65 transcription complex induced by endothelin-1 mediates signal transduction in cancer cells. Biochim. Biophys. Acta 2008, 1783, 1613–1622. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, C.; Zhong, W.; Wang, Q.; Zhang, D.; Zhang, J.; Xie, S.; Xu, M. Chrysin induces autophagy-dependent ferroptosis to increase chemosensitivity to gemcitabine by targeting CBR1 in pancreatic cancer cells. Biochem. Pharmacol. 2021, 193, 114813. [Google Scholar] [CrossRef]
- Li, Z.; Jeon, H.J.; Lee, M.J. Significance of p53, cyclin D1 and c-myc Expressions in Thyroid Tumors. Korean J. Pathol. 2004, 38, 29–34. [Google Scholar]
- Sekhon, H.S.; London, C.A.; Sekhon, M.; Iversen, P.L.; Devi, G.R. c-MYC antisense phosphosphorodiamidate morpholino oligomer inhibits lung metastasis in a murine tumor model. Lung Cancer 2008, 60, 347–354. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. Elife 2018, 7, e31770. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-Y.; Liu, H.; Zhang, J.-W.; Hu, K.; Lin, Y. Clinical significance of Smac and Ki-67 expression in pancreatic cancer. Hepatogastroenterology 2012, 59, 2640–2643. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Simal-Gandara, J.; Kopustinskiene, D.M.; Bernatoniene, J.; Samarghandian, S. Emerging cellular and molecular mechanisms underlying anticancer indications of chrysin. Cancer Cell Int. 2021, 21, 214. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar]
- Sun, Y.; Peng, W.; He, W.; Luo, M.; Chang, G.; Shen, J.; Zhao, X.; Hu, Y. Transgelin-2 is a novel target of KRAS-ERK signaling involved in the development of pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 166. [Google Scholar] [CrossRef] [Green Version]
- Skripova, V.; Vlasenkova, R.; Zhou, Y.; Astsaturov, I.; Kiyamova, R. Identification of new regulators of pancreatic cancer cell sensitivity to oxaliplatin and cisplatin. Molecules 2022, 27, 1289. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, G.S.; Moon, J.H.; Jung, J. Policosanol suppresses tumor progression in a gastric cancer xenograft model. Toxicol. Res. 2022. [Google Scholar] [CrossRef]
- Feng, H.; Gu, Z.-Y.; Li, Q.; Liu, Q.-H.; Yang, X.-Y.; Zhang, J.-J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Log (Fold Change) | p-Value |
|---|---|---|
| SRRM1 | 0.62 | 0.0170 |
| TAGLN2 | 0.64 | 0.0012 |
| FCHO2 | 0.67 | 0.0003 |
| DENND1A | 0.69 | 0.0173 |
| ZC3H18 | 0.69 | 0.0065 |
| CDH11 | 0.69 | 0.0191 |
| FASTKD2 | 0.69 | 0.0289 |
| ROCK1 | 0.70 | 0.0398 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.K.; Kwon, H.J.; Lee, G.S.; Moon, J.H.; Jung, J. Chrysin-Induced G Protein-Coupled Estrogen Receptor Activation Suppresses Pancreatic Cancer. Int. J. Mol. Sci. 2022, 23, 9673. https://doi.org/10.3390/ijms23179673
Lim HK, Kwon HJ, Lee GS, Moon JH, Jung J. Chrysin-Induced G Protein-Coupled Estrogen Receptor Activation Suppresses Pancreatic Cancer. International Journal of Molecular Sciences. 2022; 23(17):9673. https://doi.org/10.3390/ijms23179673
Chicago/Turabian StyleLim, Hyun Kyung, Hee Jung Kwon, Ga Seul Lee, Jeong Hee Moon, and Joohee Jung. 2022. "Chrysin-Induced G Protein-Coupled Estrogen Receptor Activation Suppresses Pancreatic Cancer" International Journal of Molecular Sciences 23, no. 17: 9673. https://doi.org/10.3390/ijms23179673
APA StyleLim, H. K., Kwon, H. J., Lee, G. S., Moon, J. H., & Jung, J. (2022). Chrysin-Induced G Protein-Coupled Estrogen Receptor Activation Suppresses Pancreatic Cancer. International Journal of Molecular Sciences, 23(17), 9673. https://doi.org/10.3390/ijms23179673