
Genetics behind Cerebral Disease with Ocular Comorbidity: Finding Parallels between the Brain and Eye Molecular Pathology

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. The Genetic Predisposition to CVIs

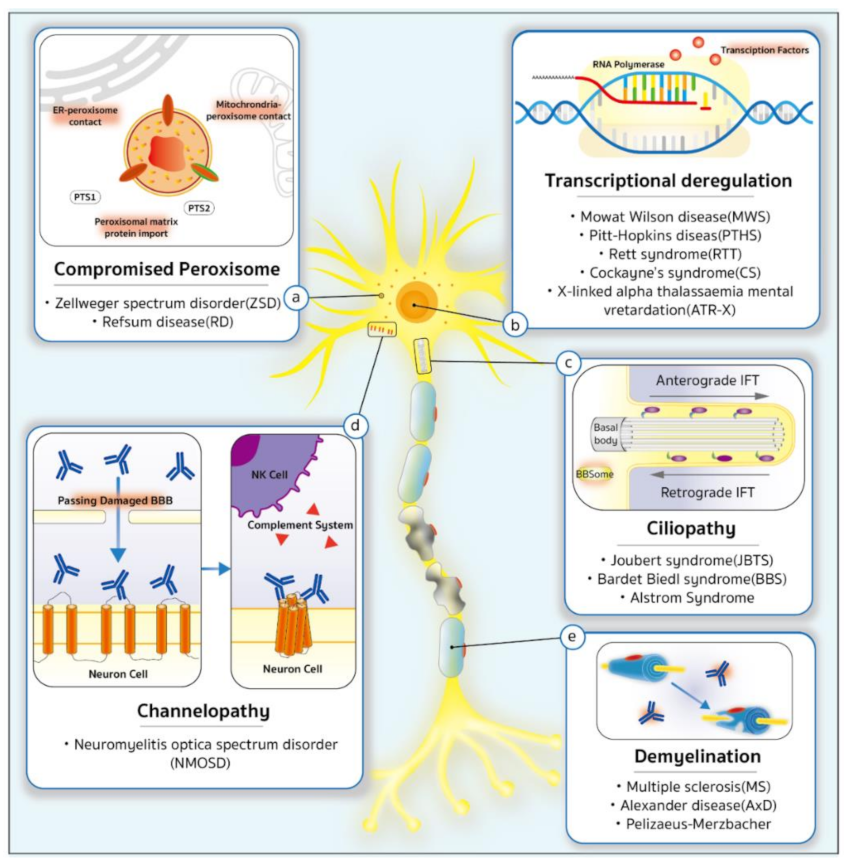{kind=link}
{kind=link}
| Type | Disorder 1 | Subtypes 2 | Age | Frequency | Male/Female Ratio | Inheritance Mode 3 | |||
|---|---|---|---|---|---|---|---|---|---|
| Onset | Diagnosed | Death (81.8 Years in General Populations [12]) | Incidence | Prevalence | |||||
| Ciliopathy | JBTS | See in Table S1 | 10 days~5 months [13] | unknown | 7.2 years [14] | 1/80,000~1/100,000 [15] | 1/80,000~1/100,000 [15,16,17] | 1.22 [18] | AR XLR (JBTS10) AD (JBTS19) |
| BBS | See in Table S2 | unknown | 9 years [19] | 25% in 44 years [20] | 1/125,000~1/160,000 in Europe population [21,22] 1/65,000 in an Arab population [23] | 1/160,000 in European population 1/13,500 in Arabic populations [24] | 1.30 [19] | AR AD (BBS1) | |
| Alstrom Syndrome | - | infancy [25] | unknown | <50 years [26] | 1/1,000,000 [27] | 1/1,000,000 [26] | 0.50 [28] | AR | |
| Demyelination | MS | - | 18 years~40 years [29] | 20 years~50 years [30] | 74.7 years [12,30] | 2.1/100,000 [31] | 35.9/100,000 [31] | 0.29~0.91 [32] | autosomal, phantom heritability |
| AxD | neonatal | <30 days [33] | unknown | <2 years [33] | 1/2,700,000 [31] | 1/2,700,000 [34] | 0.50 [28] | AD | |
| infantile | 30 days~2 years [33] | unknown | weaks~years [35] | ||||||
| juvenile | 2 years~12 years [36] | unknown | 20 years~30 years [37] | ||||||
| adult | >12 years [36] | unknown | decades [37] | ||||||
| PMD | - | 3 months~9 years [38] | unknown | 6 years~25 years [38] | 1.45/100,000~1.9/100,000 [39,40] | 1/300 000~1/500 000 [41] | >1.00 | XLR | |
| Transcriptional Deregulation | MWS | - | 27.5 months [42] | unknown | <60 years [43] | 1/70,000 [44] | 1/50,000~1/70,000 [45] | 1.00 [46,47] | AD |
| PTHS | - | 2 years~19 years [48] | unknown | unknown [49] | unknown | 1/225,000~1/300,000 [50] | 1.00 | AD | |
| RTT1 | - | 4 years [51] | 3.5 years [52] | 4 years [51] | 1/22,800 [53] | 1/10,000~1/15, 000 [53] | <1.00 [54] | XLD | |
| CS | CS type I | 0 year~2 years [55] | unknown | 16.1 years [55] | 1/200,000 [56,57] | 2.5/1,000,000 [58] | 1.00 [59] | AR | |
| CS type II | at birth [60] | unknown | 5.0 years [60] | ||||||
| CS type III | >2 years [60] | unknown | 30.3 years [60] | ||||||
| XP/CS | 0 year~2 years [61] | unknown | 7 months~6.4 years [61] | ||||||
| ATR-X | - | unknown | unknown | unknown | 1/100,000 [62] | 1/30,000~1/40,000 [63] | >1.00 | XLD | |
| Compromised Preoxisome | ZSD | - | 0 year~3.8 years [64] | 7 days~31 years [65] | depending [64] | 1/12,000 in Canadian populations 1/50,000 in US populations 1/500,000 in Japanese populations [66] | unknown | 1.00 | AR |
| RD | ARD | 2–7 years [67] | 1 year~28 years [68] | 4 decades~5 decades [69] | 1/250000 [70] | unknown | 1.00 [71] | AR | |
| IRD | early infancy [67] | unknown | 5 years~13 years [69] | ||||||
| Channelopathy | NMOSD | - | late fourth decade [72] | unknown | 52.3 years [73] | 0.053/100,000~0.400/100,00 [74] | 1/100,000 in white populations 3.5/100,000 in East Asian populations 10/100,000 in Black populations [75] | 0.11~0.43 [76,77] | Multigenic |
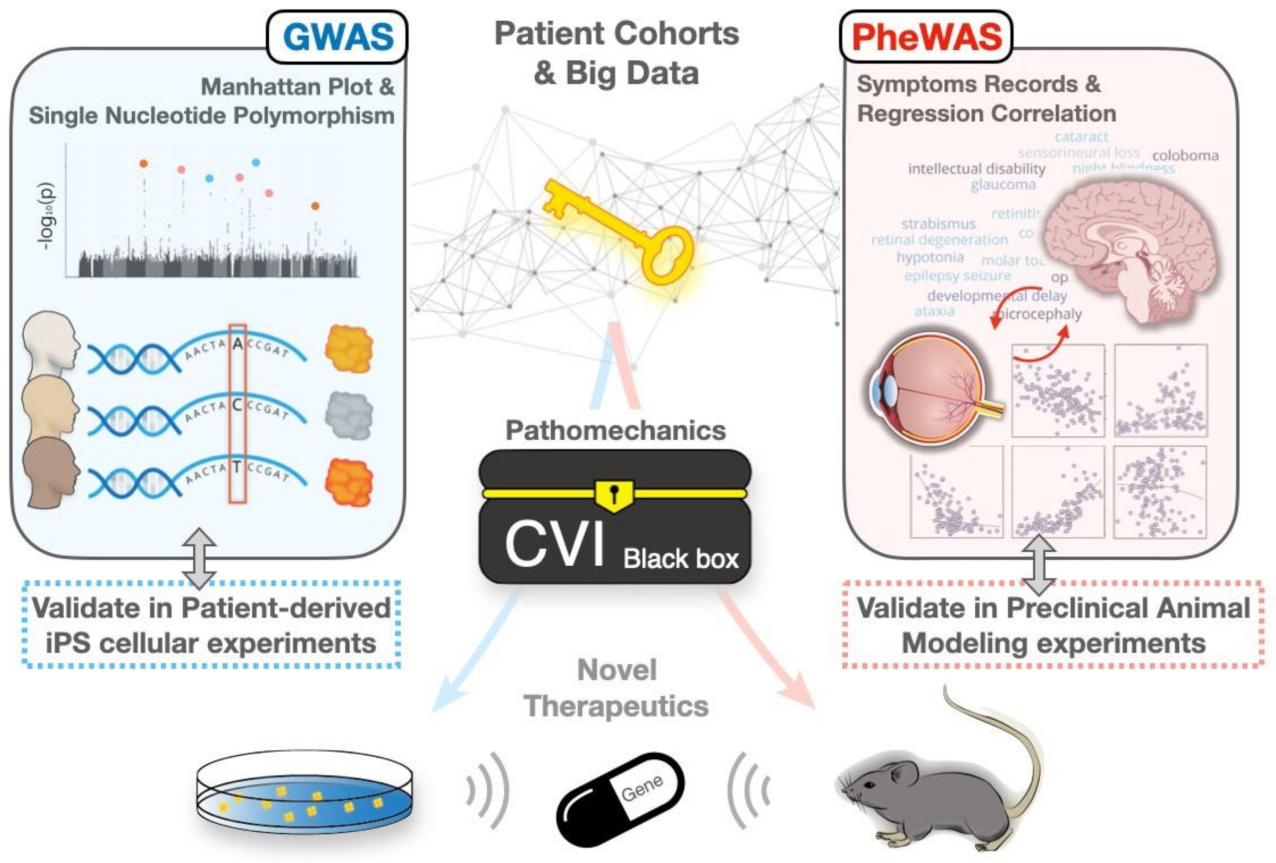
3. Revisiting CVIs by the GWAS-PheWAS Approach
4. Multiple Sclerosis: A Typical Case of Brain-Eye Parallelism
| Type | Disorder 1 | Cerebral 2 | Visual 3 |
|---|---|---|---|
| Ciliopathy | JB-Ret | MTS (100%) [194,195] developmental delay (100%) mental retardation (100%) hypotonia (100%) [196] Dandy-Walker malformation (10%) [197] | RP (100%) [198] ocular motor apraxia (80%) strabismus (74%) nystagmus (72%) [199] RD (30%) [198] chorioretinal coloboma (30%) optic nerve atrophy (22%) [199] |
| BBS | developmental delay (50–91%) ataxia (40–86%) [200] cognitive impairment (66%) central obesity (89%) [201] functional independence (74%) attention capacity (69%) [202] ID (62%) [203] perceptual reasoning (53%) verbal fluency (22–44%) [202] | RD (94%) RP (43%) [201] | |
| Alstrom Syndrome | developmental delay (45%) [204] | RD (100%) [27] blind (90%) [205] | |
| Demyelination | MS | Dawson’s fingers (92.5%) [206] central pain (15–85%) [207] central trigeminal involvement (12–38%) [208] braunstem dysfunction (25%) sensory disturbances (18.3%) motor disturbances (17.5%) [209] TN (6%) [210] ID (2%) [211] | ON (50%) [176] abnormal blink reflex (89%) [212] nystagmus (10%) [213] |
| AxD | bulbar sign (83.3%) changes in lower brain stem or upper cervical cord (82.4%) [214] cerebral white matter lesions (80%) [215] pyramidal sign (63.4%) changes in cerebellum or dentate hilum (54.1%) ataxia (50%) dysarthria (42%) [214] mental retardation (29.0%) epilepsy seizure (26.5%) pseudobulbar sign (21.6%) [216] cyst formation (25%) [217] changes in basal ganglia or thalami signal (17.6%) autonomic disturbances (11.4%) macrocephaly (9.8%) cranial sensory disturbances (6.8%) [216] | ocular motor abnormalities (46.1%) [216] nystagmus (33%) [214] | |
| PMD | developmental delay (100%) corpus callosum atrophy (100%) hypotonia (83.8%) displayed supratentorial brain atrophy (29.0%) pyramidal sign (5.4–22.2%) epilepsy seizure (7.1–14.3%) ataxia (5.4–7.4%) cerebellum atrophy (3.2%) [193] | nystagmus (99.1%) [193] | |
| Transcriptional Deregulation | MWS | hypotonia (93%) [47] microcephaly (81%) [141,218,219,220,221,222] neocortical projections (79.6%) hippocampal abnormalities (77.8%) enlargement of cerebral ventricle (68.5%) [47] epilepsy seizure (73%) [141,218,219,220,221,222] brain anomalies (43%) reduction of white matter thickness (40.7%) localized signal alterations of the white matter (22.2%) [222] | eye anomalies (4.1%) [141,223] |
| PTHS | ID (98%) gross motor development (92%) hypotonia (69%) ataxia (57%) [141] epilepsy seizure (40%) small corpus callosum (23%) enlargement of cerebral ventricle (21%) microcephaly (17%) [50] | strabismus (45%) myopia (39%) [49] astigmatism (26%) [50] nystagmus (4%) [49] | |
| RTT | deceleration of head growth (80%) epilepsy seizure (60~80%) [224] language disorder (61.5%) microcephaly (46.2%) gross motor development (30.8%) [225] | difficulty recognizing unfamiliar things [226] selectively focused on specific things [227] vision search difficulty [228] | |
| CS | abnormal myelination in brain (93%) [61] mental retardation (90%) microcephaly (83%) motor disturbance (71%) [229] tremor (66%) [190] intracranial calcifications (63%) [61] ventricular dilatation (23%) [191] epilepsy seizure (5–10%) [230] | RP (60–100%) [231] RD (33–89.3%) [58] cataracts (15–36%) [231] | |
| ATR-X | developmental delay (100%) ID (100%) [232] language disorder (95%) [233] hypotonia (80–90%) [232] microcephaly (75%) [233] brain atrophy (63%) high intensity of white matter (41%) [232] epilepsy seizure (30–40%) [232] delayed myelination (15%) [63] | ocular defects (25%) [234] | |
| Compromised Preoxisome | ZSD | peripheral neuropathy (58%) T2 hyperintensities (50%) cerebellar sign (47%) cerebellar cortical atrophy (38%) pyramidal sign (26%) high intensity of white matter (25%) hypotonia (21%) [65] | VA disability (100%) RP (84%) retinopathy (84%) night blindness (84%) retinal degeneration (63%) [65] |
| RD | polyneuropathy (70%) ataxia (50%) [192] | RP (100%) [192] pupils (78%) VA (76.7 %) visual fields (75%) cataracts (30%) nystagmus (22%) glaucoma (17%) [68] | |
| Channelopathy | NMOSD | periependymal lesions (75%) [235] central vomiting (65.38%) central hiccups (50.00%) pyramidal tract sign (42.31%) [236] LETM (32.9%) brainstem symptoms (4.5%) [237] | ON (22.4%) [238] ophthalmoplegia (19.23%) MLF syndrome (11.54%) [239] |
| Disease 1 | Brain MRI Description | Cerebral Disorders 2 | Visual Disorders 2 | Disease Process 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Epilepsy Seizure | Hypotonia | Ataxia | Microcephaly | Nystagmus | RP | RD | Cataracts | ON | |||
| Ciliopathy | ||||||||||||
| JBTS | Molar Tooth Sign (MTS) on T2 MRI | · | · | · | · |  | ||||||
| BBS | Shrinkage of the hippocampus and striatum | · | · | · | · | |||||||
| Alstrom Syndrome | Increased white matter density and small leaks near the ventricles on T1 MRI | · | ||||||||||
| Demyelinating | ||||||||||||
| MS | Finger-shaped lesion in the corpus callosum on T2 MRI | · | · | · |  | |||||||
| AxD | Shrinkage of the medulla oblongata and the upper spinal cord | · | · | · | · | |||||||
| PMD | Corpus callosum shrinkage | · | · | · | · | · | ||||||
| Transcriptional Deregulation | ||||||||||||
| MWS | Corpus callosum hypoplasia, abnormal hippocampus, ventricular enlargement | · | · | · |  | |||||||
| PTHS | Corpus callosum hypoplasia, ventricular enlargement | · | · | · | · | · | · | |||||
| RTT | Shrinkage of the corpus callosum and the cerebellum, brainstem narrowing | · | · | · | ||||||||
| CS | Calcification | · | · | · | · | · | ||||||
| ATR-X | Brain shrinkage, ventricular enlargement | · | · | · | ||||||||
| Compromised Peroxisome | ||||||||||||
| ZSD | T2 hyperintensity | · | · | · |  | |||||||
| RD | Increased white matter density near the ventricles on T2 MRI | · | · | · | · | |||||||
| Channelopathy | ||||||||||||
| NMOSD | Marbled lesions above the corpus callosum | · | · | |||||||||
5. Ciliopathy
5.1. Joubert Syndrome
5.2. Bardet–Biedl Syndrome
5.3. Alstrom Syndrome
6. Demyelinating Diseases
6.1. Alexander Disease
6.2. Pelizaeus–Merzbacher Disease
6.3. Possible Treatments
7. Transcriptional Deregulation
7.1. Mowat–Wilson Syndrome
7.2. Pitt–Hopkins Syndrome
7.3. Rett Syndrome
7.4. Cockayne Syndrome
7.5. X-linked Alpha Thalassaemia Mental Retardation
8. Compromised Peroxisomes
8.1. Zellweger Spectrum Disorder
8.2. Refsum Disease
9. Channelopathies
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boonstra, N.; Limburg, H.; Tijmes, N.; van Genderen, M.; Schuil, J.; van Nispen, R. Changes in causes of low vision between 1988 and 2009 in a Dutch population of children. Acta Ophthalmol. 2012, 90, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, N.M.; Jackson, A.J.; Hill, A.E. Visual impairment in childhood: Insights from a community-based survey. Child. Care Health Dev. 2003, 29, 493–499. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Saunders, K.J.; Little, J.A. What assessments are currently used to investigate and diagnose cerebral visual impairment (CVI) in children? A systematic review. Ophthalmic Physiol. Opt. 2021, 41, 224–244. [Google Scholar] [CrossRef]
- Lueck, A.H.; Dutton, G.N.; Chokron, S. Profiling Children With Cerebral Visual Impairment Using Multiple Methods of Assessment to Aid in Differential Diagnosis. Semin. Pediatr. Neurol. 2019, 31, 5–14. [Google Scholar] [CrossRef]
- Surguchev, A.; Surguchov, A. Conformational diseases: Looking into the eyes. Brain Res. Bull. 2010, 81, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Maurage, C.A.; Ruchoux, M.M.; De Vos, R.; Surguchov, A.; Destee, A. Retinal involvement in dementia with Lewy bodies: A clue to hallucinations? Ann. Neurol. 2003, 54, 542–547. [Google Scholar] [CrossRef]
- Jonsson, H.; Sulem, P.; Kehr, B.; Kristmundsdottir, S.; Zink, F.; Hjartarson, E.; Hardarson, M.T.; Hjorleifsson, K.E.; Eggertsson, H.P.; Gudjonsson, S.A.; et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 2017, 549, 519–522. [Google Scholar] [CrossRef]
- Junemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; von Haeseler, A.; Stoye, J.; et al. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013, 31, 294–296. [Google Scholar] [CrossRef]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar] [CrossRef]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef]
- Povysil, G.; Heinzl, M.; Salazar, R.; Stoler, N.; Nekrutenko, A.; Tiemann-Boege, I. Erratum: Increased yields of duplex sequencing data by a series of quality control tools. NAR Genom. Bioinform. 2021, 3, lqab014. [Google Scholar] [CrossRef] [PubMed]
- Lunde, H.M.B.; Assmus, J.; Myhr, K.M.; Bo, L.; Grytten, N. Survival and cause of death in multiple sclerosis: A 60-year longitudinal population study. J. Neurol. Neurosurg. Psychiatry 2017, 88, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Elhassanien, A.F.; Alghaiaty, H.A. Joubert syndrome: Clinical and radiological characteristics of nine patients. Ann. Indian Acad. Neurol. 2013, 16, 239–244. [Google Scholar] [CrossRef]
- Dempsey, J.C.; Phelps, I.G.; Bachmann-Gagescu, R.; Glass, I.A.; Tully, H.M.; Doherty, D. Mortality in Joubert syndrome. Am. J. Med. Genet. A 2017, 173, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef]
- Kroes, H.Y.; Monroe, G.R.; van der Zwaag, B.; Duran, K.J.; de Kovel, C.G.; van Roosmalen, M.J.; Harakalova, M.; Nijman, I.J.; Kloosterman, W.P.; Giles, R.H.; et al. Joubert syndrome: Genotyping a Northern European patient cohort. Eur. J. Hum. Genet. 2016, 24, 214–220. [Google Scholar] [CrossRef]
- Phelps, I.G.; Dempsey, J.C.; Grout, M.E.; Isabella, C.R.; Tully, H.M.; Doherty, D.; Bachmann-Gagescu, R. Interpreting the clinical significance of combined variants in multiple recessive disease genes: Systematic investigation of Joubert syndrome yields little support for oligogenicity. Genet. Med. 2018, 20, 223–233. [Google Scholar] [CrossRef]
- Nuovo, S.; Bacigalupo, I.; Ginevrino, M.; Battini, R.; Bertini, E.; Borgatti, R.; Casella, A.; Micalizzi, A.; Nardella, M.; Romaniello, R.; et al. Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology 2020, 94, e797–e801. [Google Scholar] [CrossRef]
- Beales, P.; Elcioglu, N.; Woolf, A.; Parker, D.; Flinter, F. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef]
- O’Dea, D.; Parfrey, P.S.; Harnett, J.D.; Hefferton, D.; Cramer, B.C.; Green, J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am. J. Kidney Dis. 1996, 27, 776–783. [Google Scholar] [CrossRef]
- Klein, D.; Ammann, F. The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland: Clinical, genetic and epidemiological studies. J. Neurol. Sci. 1969, 9, 479–513. [Google Scholar] [CrossRef]
- Beales, P.L.; Warner, A.M.; Hitman, G.A.; Thakker, R.; Flinter, F.A. Bardet-Biedl syndrome: A molecular and phenotypic study of 18 families. J. Med. Genet. 1997, 34, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Farag, T.I.; Teebi, A.S. Bardet-Biedl and Laurence-Moon syndromes in a mixed Arab population. Clin. Genet. 1988, 33, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Marshall, J.D.; Bronson, R.T.; Collin, G.B.; Nordstrom, A.D.; Maffei, P.; Paisey, R.B.; Carey, C.; Macdermott, S.; Russell-Eggitt, I.; Shea, S.E.; et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch. Intern. Med. 2005, 165, 675–683. [Google Scholar] [CrossRef]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alstrom syndrome: Genetics and clinical overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef]
- Tahani, N.; Maffei, P.; Dollfus, H.; Paisey, R.; Valverde, D.; Milan, G.; Han, J.C.; Favaretto, F.; Madathil, S.C.; Dawson, C.; et al. Consensus clinical management guidelines for Alstrom syndrome. Orphanet J. Rare Dis. 2020, 15, 253. [Google Scholar] [CrossRef]
- Jacob, J.; Robertson, N.J.; Hilton, D.A. The clinicopathological spectrum of Rosenthal fibre encephalopathy and Alexander’s disease: A case report and review of the literature. J. Neurol. Neurosurg. Psychiatry 2003, 74, 807–810. [Google Scholar] [CrossRef]
- Paty, D.W.; Boiko, A.N.; Vorobeychi, G. Multiple sclerosis with early and late disease onset. In Blue Books of Practical Neurology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 27, pp. 285–302. [Google Scholar]
- Midgard, R.; Albrektsen, G.; Riise, T.; Kvåle, G.; Nyland, H. Prognostic factors for survival in multiple sclerosis: A longitudinal, population based study in Møre and Romsdal, Norway. J. Neurology. Neurosurg. Psychiatry 1995, 58, 417–421. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Bostrom, I.; Stawiarz, L.; Landtblom, A.M. Sex ratio of multiple sclerosis in the National Swedish MS Register (SMSreg). Mult. Scler. 2013, 19, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Waldman, A.; Naidu, S. Alexander Disease. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Yoshida, T.; Sasaki, M.; Yoshida, M.; Namekawa, M.; Okamoto, Y.; Tsujino, S.; Sasayama, H.; Mizuta, I.; Nakagawa, M.; Alexander Disease Study Group in Japan. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol. 2011, 258, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Bassuk, A.G.; Joshi, A.; Burton, B.K.; Larsen, M.B.; Burrowes, D.M.; Stack, C. Alexander disease with serial MRS and a new mutation in the glial fibrillary acidic protein gene. Neurology 2003, 61, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Fancellu, R.; Mariotti, C.; Romano, S.; Salmaggi, A.; Carella, F.; Girotti, F.; Gattellaro, G.; Carriero, M.R.; Farina, L.; et al. Adult-onset Alexander disease: A series of eleven unrelated cases with review of the literature. Brain 2008, 131 Pt 9, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Cascella, M. Alexander Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Goldman, L.; Schafer, A.I. Goldman-Cecil Medicine E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Bonkowsky, J.L.; Nelson, C.; Kingston, J.L.; Filloux, F.M.; Mundorff, M.B.; Srivastava, R. The burden of inherited leukodystrophies in children. Neurology 2010, 75, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Numata, Y.; Gotoh, L.; Iwaki, A.; Kurosawa, K.; Takanashi, J.; Deguchi, K.; Yamamoto, T.; Osaka, H.; Inoue, K. Epidemiological, clinical, and genetic landscapes of hypomyelinating leukodystrophies. J. Neurol. 2014, 261, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wang, L. Pelizaeus-Merzbacher disease: Molecular diagnosis and therapy. Intractable Rare Dis. Res. 2013, 2, 103–105. [Google Scholar] [CrossRef][Green Version]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef]
- Adam, M.P.; Conta, J.; Bean, L.J.H. Mowat-Wilson Syndrome. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mowat, D.; Wilson, M. Mowat-Wilson syndrome. In Cassidy and Allanson’s Management of Genetic Syndromes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 597–609. [Google Scholar]
- Cassidy, S.B.; Allanson, J.E. Management of Genetic Syndromes, 3rd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2010; p. xxii. 962p. [Google Scholar]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef]
- Garavelli, L.; Mainardi, P.C. Mowat-Wilson syndrome. Orphanet J. Rare Dis. 2007, 2, 42. [Google Scholar] [CrossRef]
- Pitt, D.; Hopkins, I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. J. Paediatr. Child Health 1978, 14, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Peippo, M.; Ignatius, J. Pitt-Hopkins Syndrome. Mol. Syndromol. 2012, 2, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Zweier, C.; Van Balkom, I.D.; Sweetser, D.A.; Alaimo, J.; Bijlsma, E.K.; Cody, J.; Elsea, S.H.; Giurgea, I.; Macchiaiolo, M.; et al. Diagnosis and management in Pitt-Hopkins syndrome: First international consensus statement. Clin. Genet. 2019, 95, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Jian, L.; Nagarajan, L.; de Klerk, N.; Ravine, D.; Bower, C.; Anderson, A.; Williamson, S.; Christodoulou, J.; Leonard, H. Predictors of seizure onset in Rett syndrome. J. Pediatr. 2006, 149, 542–547. [Google Scholar] [CrossRef]
- Fehr, S.; Bebbington, A.; Nassar, N.; Downs, J.; Ronen, G.M.; Leonard, H. Trends in the diagnosis of Rett syndrome in Australia. Pediatr. Res. 2011, 70, 313–319. [Google Scholar] [CrossRef]
- Chahil, G.; Bollu, P.C. Rett Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Reichow, B.; George-Puskar, A.; Lutz, T.; Smith, I.C.; Volkmar, F.R. Brief report: Systematic review of Rett syndrome in males. J. Autism Dev. Disord. 2015, 45, 3377–3383. [Google Scholar] [CrossRef]
- Natale, V. A comprehensive description of the severity groups in Cockayne syndrome. Am. J. Med. Genet. A 2011, 155A, 1081–1095. [Google Scholar] [CrossRef]
- Pascucci, B.; Fragale, A.; Marabitti, V.; Leuzzi, G.; Calcagnile, A.S.; Parlanti, E.; Franchitto, A.; Dogliotti, E.; D’Errico, M. CSA and CSB play a role in the response to DNA breaks. Oncotarget 2018, 9, 11581–11591. [Google Scholar] [CrossRef]
- Pines, A.; Dijk, M.; Makowski, M.; Meulenbroek, E.M.; Vrouwe, M.G.; van der Weegen, Y.; Baltissen, M.; French, P.J.; van Royen, M.E.; Luijsterburg, M.S.; et al. TRiC controls transcription resumption after UV damage by regulating Cockayne syndrome protein A. Nat. Commun. 2018, 9, 1040. [Google Scholar] [CrossRef]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef]
- Ataee, P.; Karimi, A.; Eftekhari, K. Hepatic Failure following Metronidazole in Children with Cockayne Syndrome. Case Rep. Pediatr. 2020, 2020, 9634196. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Natale, V.; Raquer, H. Xeroderma pigmentosum-Cockayne syndrome complex. Orphanet J. Rare Dis. 2017, 12, 65. [Google Scholar] [CrossRef]
- Villard, L.; Fontes, M. Alpha-thalassemia/mental retardation syndrome, X-Linked (ATR-X, MIM #301040, ATR-X/XNP/XH2 gene MIM #300032). Eur. J. Hum. Genet. 2002, 10, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Ban, H.; Matsufuji, M.; Okamoto, N.; Enomoto, K.; Kurosawa, K.; Aida, N. Neuroradiologic features in X-linked α-thalassemia/mental retardation syndrome. AJNR Am. J. Neuroradiol. 2013, 34, 2034–2038. [Google Scholar] [CrossRef]
- Bose, M.; Yergeau, C.; D’Souza, Y.; Cuthbertson, D.D.; Lopez, M.J.; Smolen, A.K.; Braverman, N.E. Characterization of Severity in Zellweger Spectrum Disorder by Clinical Findings: A Scoping Review, Meta-Analysis and Medical Chart Review. Cells 2022, 11, 1891. [Google Scholar] [CrossRef] [PubMed]
- Berendse, K.; Engelen, M.; Ferdinandusse, S.; Majoie, C.B.; Waterham, H.R.; Vaz, F.M.; Koelman, J.H.; Barth, P.G.; Wanders, R.J.; Poll-The, B.T. Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 2016, 39, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, V.; Pasrija, D. Zellweger Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Van den Brink, D.M.; Brites, P.; Haasjes, J.; Wierzbicki, A.S.; Mitchell, J.; Lambert-Hamill, M.; de Belleroche, J.; Jansen, G.A.; Waterham, H.R.; Wanders, R.J. Identification of PEX7 as the second gene involved in Refsum disease. Adv. Exp. Med. Biol. 2003, 544, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Claridge, K.G.; Gibberd, F.B.; Sidey, M.C. Refsum disease: The presentation and ophthalmic aspects of Refsum disease in a series of 23 patients. Eye 1992, 6 Pt 4, 371–375. [Google Scholar] [CrossRef]
- Kumar, R.; De Jesus, O. Refsum Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Jayaram, H.; Downes, S.M. Midlife diagnosis of Refsum disease in siblings with retinitis pigmentosa—The footprint is the clue: A case report. J. Med. Case Rep. 2008, 2, 80. [Google Scholar] [CrossRef]
- Richterich, R.; Rosin, S.; Rossi, E. Refsum’s disease (heredopathia atactica polyneuritiformis). An inborn error of lipid metabolism with storage of 3,7,11,15 tetramethyl hexadecanoic acid formal genetics. Humangenetik 1965, 1, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M. Neuromyelitis optica: Effect of gender. J. Neurol. Sci. 2009, 286, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Mealy, M.A.; Kessler, R.A.; Rimler, Z.; Reid, A.; Totonis, L.; Cutter, G.; Kister, I.; Levy, M. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e468. [Google Scholar] [CrossRef]
- Marrie, R.A.; Gryba, C. The incidence and prevalence of neuromyelitis optica: A systematic review. Int. J. MS Care 2013, 15, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.Y.; Asgari, N.; Nakashima, I.; Broadley, S.A.; Leite, M.I.; Kissani, N.; Jacob, A.; Marignier, R.; Weinshenker, B.G.; Paul, F.; et al. Epidemiology of Neuromyelitis Optica Spectrum Disorder and Its Prevalence and Incidence Worldwide. Front. Neurol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef]
- McKeon, A.; Lennon, V.A.; Lotze, T.; Tenenbaum, S.; Ness, J.M.; Rensel, M.; Kuntz, N.L.; Fryer, J.P.; Homburger, H.; Hunter, J.; et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008, 71, 93–100. [Google Scholar] [CrossRef]
- Ozaki, K.; Ohnishi, Y.; Iida, A.; Sekine, A.; Yamada, R.; Tsunoda, T.; Sato, H.; Sato, H.; Hori, M.; Nakamura, Y.; et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 2002, 32, 650–654. [Google Scholar] [CrossRef]
- Fitzgerald, K.C.; Kim, K.; Smith, M.D.; Aston, S.A.; Fioravante, N.; Rothman, A.M.; Krieger, S.; Cofield, S.S.; Kimbrough, D.J.; Bhargava, P.; et al. Early complement genes are associated with visual system degeneration in multiple sclerosis. Brain 2019, 142, 2722–2736. [Google Scholar] [CrossRef]
- Watanabe, M.; Nakamura, Y.; Sato, S.; Niino, M.; Fukaura, H.; Tanaka, M.; Ochi, H.; Kanda, T.; Takeshita, Y.; Yokota, T.; et al. HLA genotype-clinical phenotype correlations in multiple sclerosis and neuromyelitis optica spectrum disorders based on Japan MS/NMOSD Biobank data. Sci. Rep. 2021, 11, 607. [Google Scholar] [CrossRef]
- Denny, J.C.; Ritchie, M.D.; Basford, M.A.; Pulley, J.M.; Bastarache, L.; Brown-Gentry, K.; Wang, D.; Masys, D.R.; Roden, D.M.; Crawford, D.C. PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010, 26, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. 2013, 29, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Barnard, B.; Sussman, M.; Bondurant, S.S.; Nienhuis, J.; Krysan, P. Microarrays (DNA chips) for the classroom laboratory. Biochem. Mol. Biol. Educ. 2006, 34, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef] [PubMed]
- Bodi, K.; Perera, A.G.; Adams, P.S.; Bintzler, D.; Dewar, K.; Grove, D.S.; Kieleczawa, J.; Lyons, R.H.; Neubert, T.A.; Noll, A.C.; et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J. Biomol. Tech. 2013, 24, 73–86. [Google Scholar] [CrossRef]
- Hayden, E.C. Technology: The $1000 genome. Nature 2014, 507, 294–295. [Google Scholar] [CrossRef]
- Schaller, R.R. Moore’s law: Past, present and future. IEEE Spectr. 1997, 34, 52–59. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attie-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Mégarbané, A.; Hmaimess, G.; Bizzari, S.; El-Bazzal, L.; Al-Ali, M.T.; Stora, S.; El-Hayek, S. A novel PDE6D mutation in a patient with Joubert syndrome type 22 (JBTS22). Eur. J. Med. Genet. 2019, 62, 103576. [Google Scholar] [CrossRef]
- Lee, J.E.; Silhavy, J.L.; Zaki, M.S.; Schroth, J.; Bielas, S.L.; Marsh, S.E.; Olvera, J.; Brancati, F.; Iannicelli, M.; Ikegami, K.; et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 2012, 44, 193–199. [Google Scholar] [CrossRef]
- Thomas, S.; Wright, K.J.; Le Corre, S.; Micalizzi, A.; Romani, M.; Abhyankar, A.; Saada, J.; Perrault, I.; Amiel, J.; Litzler, J.; et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat. 2014, 35, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Hamdan, F.F.; McKnight, D.; Davis, E.; Mandel, H.; Schwartzentruber, J.; Martin, B.; Patry, L.; Nassif, C.; Dionne-Laporte, A. Joubert syndrome in French Canadians and identification of mutations in CEP104. Am. J. Hum. Genet. 2015, 97, 744–753. [Google Scholar] [CrossRef]
- Cauley, E.S.; Hamed, A.; Mohamed, I.N.; Elseed, M.; Martinez, S.; Yahia, A.; Abozar, F.; Abubakr, R.; Koko, M.; Elsayed, L. Overlap of polymicrogyria, hydrocephalus, and Joubert syndrome in a family with novel truncating mutations in ADGRG1/GPR56 and KIAA0556. Neurogenetics 2019, 20, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Micalizzi, A.; Kraoua, I.; Dotti, M.T.; Cavallin, M.; Sztriha, L.; Ruta, R.; Mancini, F.; Mazza, T.; Castellana, S.; et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: Expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J. Rare Dis. 2014, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.H.; Phelps, I.G.; et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am. J. Hum. Genet. 2017, 101, 23–36. [Google Scholar] [CrossRef]
- De Mori, R.; Romani, M.; D’Arrigo, S.; Zaki, M.S.; Lorefice, E.; Tardivo, S.; Biagini, T.; Stanley, V.; Musaev, D.; Fluss, J.; et al. Hypomorphic Recessive Variants in SUFU Impair the Sonic Hedgehog Pathway and Cause Joubert Syndrome with Cranio-facial and Skeletal Defects. Am. J. Hum. Genet. 2017, 101, 552–563. [Google Scholar] [CrossRef]
- Satoda, Y.; Noguchi, T.; Fujii, T.; Taniguchi, A.; Katoh, Y.; Nakayama, K. BROMI/TBC1D32 together with CCRK/CDK20 and FAM149B1/JBTS36 contributes to IFT turnaround involving ICK/CILK1. Mol. Biol. Cell 2022, 33, 9. [Google Scholar] [CrossRef]
- Alkanderi, S.; Molinari, E.; Shaheen, R.; Elmaghloob, Y.; Stephen, L.A.; Sammut, V.; Ramsbottom, S.A.; Srivastava, S.; Cairns, G.; Edwards, N.; et al. ARL3 Mutations Cause Joubert Syndrome by Disrupting Ciliary Protein Composition. Am. J. Hum. Genet. 2018, 103, 612–620. [Google Scholar] [CrossRef]
- Shaheen, R.; Jiang, N.; Alzahrani, F.; Ewida, N.; Al-Sheddi, T.; Alobeid, E.; Musaev, D.; Stanley, V.; Hashem, M.; Ibrahim, N.; et al. Bi-allelic Mutations in FAM149B1 Cause Abnormal Primary Cilium and a Range of Ciliopathy Phenotypes in Humans. Am. J. Hum. Genet. 2019, 104, 731–737. [Google Scholar] [CrossRef]
- Latour, B.L.; Van De Weghe, J.C.; Rusterholz, T.D.; Letteboer, S.J.; Gomez, A.; Shaheen, R.; Gesemann, M.; Karamzade, A.; Asadollahi, M.; Barroso-Gil, M.; et al. Dysfunction of the ciliary ARMC9/TOGARAM1 protein module causes Joubert syndrome. J. Clin. Investig. 2020, 130, 4423–4439. [Google Scholar] [CrossRef]
- Stephen, J.; Vilboux, T.; Mian, L.; Kuptanon, C.; Sinclair, C.M.; Yildirimli, D.; Maynard, D.M.; Bryant, J.; Fischer, R.; Vemulapalli, M.; et al. Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum. Genet. 2017, 136, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Van De Weghe, J.C.; Giordano, J.L.; Mathijssen, I.B.; Mojarrad, M.; Lugtenberg, D.; Miller, C.V.; Dempsey, J.C.; Mohajeri, M.S.A.; van Leeuwen, E.; Pajkrt, E.; et al. TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes. HGG Adv. 2021, 2, 100016. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Lin, Z.; Zhu, T.; Jin, M.; Meng, D.; He, R.; Cao, Z.; Shen, Y.; Lu, C.; Cai, R.; et al. Disrupted intraflagellar transport due to IFT74 variants causes Joubert syndrome. Genet. Med. 2021, 23, 1041–1049. [Google Scholar] [CrossRef]
- Bielas, S.L.; Silhavy, J.L.; Brancati, F.; Kisseleva, M.V.; Al-Gazali, L.; Sztriha, L.; Bayoumi, R.A.; Zaki, M.S.; Abdel-Aleem, A.; Rosti, R.O.; et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009, 41, 1032–1036. [Google Scholar] [CrossRef]
- Valente, E.M.; Logan, C.V.; Mougou-Zerelli, S.; Lee, J.H.; Silhavy, J.L.; Brancati, F.; Iannicelli, M.; Travaglini, L.; Romani, S.; Illi, B.; et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010, 42, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Ferland, R.J.; Eyaid, W.; Collura, R.V.; Tully, L.D.; Hill, R.S.; Al-Nouri, D.; Al-Rumayyan, A.; Topcu, M.; Gascon, G.; Bodell, A.; et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 2004, 36, 1008–1013. [Google Scholar] [CrossRef]
- Parisi, M.A.; Bennett, C.L.; Eckert, M.L.; Dobyns, W.B.; Gleeson, J.G.; Shaw, D.W.; McDonald, R.; Eddy, A.; Chance, P.F.; Glass, I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004, 75, 82–91. [Google Scholar] [CrossRef]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.; Letteboer, S.J.; van Beersum, S.E.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef]
- Valente, E.M.; Brancati, F.; Boltshauser, E.; Dallapiccola, B. Clinical utility gene card for: Joubert Syndrome—update 2013. Eur. J. Hum. Genet. 2013, 21, 1187. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Silhavy, J.L.; Lee, J.E.; Al-Gazali, L.; Thomas, S.; Davis, E.E.; Bielas, S.L.; Hill, K.J.; Iannicelli, M.; Brancati, F.; et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 2012, 335, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Hamdan, F.F.; Schwartzentruber, J.A.; Patry, L.; Ospina, L.H.; Shevell, M.I.; Desilets, V.; Dobrzeniecka, S.; Mathonnet, G.; Lemyre, E.; et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J. Med. Genet. 2012, 49, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Lambacher, N.J.; Bruel, A.L.; van Dam, T.J.; Szymanska, K.; Slaats, G.G.; Kuhns, S.; McManus, G.J.; Kennedy, J.E.; Gaff, K.; Wu, K.M.; et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol. 2016, 18, 122–131. [Google Scholar] [CrossRef]
- Baala, L.; Romano, S.; Khaddour, R.; Saunier, S.; Smith, U.M.; Audollent, S.; Ozilou, C.; Faivre, L.; Laurent, N.; Foliguet, B.; et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007, 80, 186–194. [Google Scholar] [CrossRef]
- Coene, K.L.; Roepman, R.; Doherty, D.; Afroze, B.; Kroes, H.Y.; Letteboer, S.J.; Ngu, L.H.; Budny, B.; van Wijk, E.; Gorden, N.T.; et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 2009, 85, 465–481. [Google Scholar] [CrossRef]
- Dafinger, C.; Liebau, M.C.; Elsayed, S.M.; Hellenbroich, Y.; Boltshauser, E.; Korenke, G.C.; Fabretti, F.; Janecke, A.R.; Ebermann, I.; Nurnberg, G.; et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Investig. 2011, 121, 2662–2667. [Google Scholar] [CrossRef]
- Srour, M.; Schwartzentruber, J.; Hamdan, F.F.; Ospina, L.H.; Patry, L.; Labuda, D.; Massicotte, C.; Dobrzeniecka, S.; Capo-Chichi, J.M.; Papillon-Cavanagh, S.; et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet. 2012, 90, 693–700. [Google Scholar] [CrossRef]
- Thomas, S.; Legendre, M.; Saunier, S.; Bessieres, B.; Alby, C.; Bonniere, M.; Toutain, A.; Loeuillet, L.; Szymanska, K.; Jossic, F.; et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet. 2012, 91, 372–378. [Google Scholar] [CrossRef]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef]
- Shaheen, R.; Shamseldin, H.E.; Loucks, C.M.; Seidahmed, M.Z.; Ansari, S.; Ibrahim Khalil, M.; Al-Yacoub, N.; Davis, E.E.; Mola, N.A.; Szymanska, K.; et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet. 2014, 94, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Akizu, N.; Silhavy, J.L.; Rosti, R.O.; Scott, E.; Fenstermaker, A.G.; Schroth, J.; Zaki, M.S.; Sanchez, H.; Gupta, N.; Kabra, M.; et al. Mutations in CSPP1 lead to classical Joubert syndrome. Am. J. Hum. Genet. 2014, 94, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Tuz, K.; Bachmann-Gagescu, R.; O’Day, D.R.; Hua, K.; Isabella, C.R.; Phelps, I.G.; Stolarski, A.E.; O’Roak, B.J.; Dempsey, J.C.; Lourenco, C.; et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2014, 94, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; Phelps, I.G.; Dempsey, J.C.; Sharma, V.A.; Ishak, G.E.; Boyle, E.A.; Wilson, M.; Marques Lourenco, C.; Arslan, M.; University of Washington Center for Mendelian, G.; et al. KIAA0586 is Mutated in Joubert Syndrome. Hum. Mutat. 2015, 36, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Hofree, M.; Kim, S.; Scott, E.; Copeland, B.; Romani, M.; Silhavy, J.L.; Rosti, R.O.; Schroth, J.; Mazza, T.; et al. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. Elife 2015, 4, e06602. [Google Scholar] [CrossRef]
- Malicdan, M.C.; Vilboux, T.; Stephen, J.; Maglic, D.; Mian, L.; Konzman, D.; Guo, J.; Yildirimli, D.; Bryant, J.; Fischer, R.; et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J. Med. Genet. 2015, 52, 830–839. [Google Scholar] [CrossRef]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef]
- Shaheen, R.; Schmidts, M.; Faqeih, E.; Hashem, A.; Lausch, E.; Holder, I.; Superti-Furga, A.; Consortium, U.K.; Mitchison, H.M.; Almoisheer, A.; et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum. Mol. Genet. 2015, 24, 1410–1419. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Nishimura, D.Y.; Searby, C.C.; Shastri, M.; Yen, H.J.; Beck, J.S.; Braun, T.; Streb, L.M.; Cornier, A.S.; Cox, G.F.; et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 2002, 31, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Florea, L.; Caba, L.; Gorduza, E.V. Bardet-Biedl Syndrome-Multiple Kaleidoscope Images: Insight into Mechanisms of Genotype-Phenotype Correlations. Genes 2021, 12, 1353. [Google Scholar] [CrossRef]
- Marshall, J.D.; Hinman, E.G.; Collin, G.B.; Beck, S.; Cerqueira, R.; Maffei, P.; Milan, G.; Zhang, W.; Wilson, D.I.; Hearn, T.; et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum. Mutat. 2007, 28, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Moutsianas, L.; Jostins, L.; Beecham, A.H.; Dilthey, A.T.; Xifara, D.K.; Ban, M.; Shah, T.S.; Patsopoulos, N.A.; Alfredsson, L.; Anderson, C.A.; et al. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics, C. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflammation 2020, 17, 260. [Google Scholar] [CrossRef]
- Iwaki, T.; Kume-Iwaki, A.; Liem, R.K.; Goldman, J.E. αB-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell 1989, 57, 71–78. [Google Scholar] [CrossRef]
- Johnson, A.B.; Bettica, A. On-grid immunogold labeling of glial intermediate filaments in epoxy-embedded tissue. Am. J. Anat. 1989, 185, 335–341. [Google Scholar] [CrossRef]
- Inoue, K.; Osaka, H.; Thurston, V.C.; Clarke, J.T.; Yoneyama, A.; Rosenbarker, L.; Bird, T.D.; Hodes, M.E.; Shaffer, L.G.; Lupski, J.R. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am. J. Hum. Genet. 2002, 71, 838–853. [Google Scholar] [CrossRef]
- Dastot-Le Moal, F.; Wilson, M.; Mowat, D.; Collot, N.; Niel, F.; Goossens, M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum. Mutat. 2007, 28, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Hellwig, M.; Harrison, L.; Holdhof, D.; Lauffer, M.C.; Niesen, J.; Virdi, S.; Indenbirken, D.; Schuller, U. The basic helix-loop-helix transcription factor TCF4 impacts brain architecture as well as neuronal morphology and differentiation. Eur. J. Neurosci. 2020, 51, 2219–2235. [Google Scholar] [CrossRef]
- Thambirajah, A.A.; Ng, M.K.; Frehlick, L.J.; Li, A.; Serpa, J.J.; Petrotchenko, E.V.; Silva-Moreno, B.; Missiaen, K.K.; Borchers, C.H.; Adam Hall, J.; et al. MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 2012, 40, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. The many faces of Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15273–15274. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, M.; Fawcett, H.; Botta, E.; Nardo, T.; Lehmann, A.R. Genetic analysis of twenty-two patients with Cockayne syndrome. Hum.Genet. 1996, 97, 418–423. [Google Scholar] [CrossRef]
- Garrick, D.; Samara, V.; McDowell, T.L.; Smith, A.J.; Dobbie, L.; Higgs, D.R.; Gibbons, R.J. A conserved truncated isoform of the ATR-X syndrome protein lacking the SWI/SNF-homology domain. Gene 2004, 326, 23–34. [Google Scholar] [CrossRef]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta 2006, 1763, 1733–1748. [Google Scholar] [CrossRef]
- Nanetti, L.; Pensato, V.; Leoni, V.; Rizzetto, M.; Caccia, C.; Taroni, F.; Mariotti, C.; Gellera, C. PEX7 mutations cause congenital cataract retinopathy and late-onset ataxia and cognitive impairment: Report of two siblings and review of the literature. J. Clin. Neurol. 2015, 11, 197–199. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Li, Y.; Dong, S.A.; Yi, M.; Zhang, Q.X.; Feng, B.; Yang, L.; Shi, F.D.; Yang, C.S. Multi-Level Analyses of Genome-Wide Association Study to Reveal Significant Risk Genes and Pathways in Neuromyelitis Optica Spectrum Disorder. Front. Genet. 2021, 12, 690537. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, C.; Sun, X.; Wang, J.; Li, R.; Chang, Y.; Fan, P.; Wang, Y.; Wu, Y.; Peng, L.; et al. Whole-exome sequencing reveals the major genetic factors contributing to neuromyelitis optica spectrum disorder in Chinese patients with aquaporin 4-IgG seropositivity. Eur. J. Neurol. 2021, 28, 2294–2304. [Google Scholar] [CrossRef]
- Palterer, B.; Brugnolo, F.; Sieni, E.; Barilaro, A.; Parronchi, P. Neuromyelitis optica, atypical hemophagocytic lymphohistiocytosis and heterozygous perforin A91V mutation. J. Neuroimmunol. 2017, 311, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.P.; Wang, H.X.; Zhuang, J.C.; Liu, Q.B.; Zhao, G.X.; Li, Z.X.; Wu, Z.Y. Variants of autophagy-related gene 5 are associated with neuromyelitis optica in the Southern Han Chinese population. Autoimmunity 2014, 47, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, H.Y.; Kim, E.; Lee, K.S.; Kim, K.K.; Choi, B.O.; Kim, S.M.; Bae, J.S.; Lee, S.O.; Chun, J.Y.; et al. Common CYP7A1 promoter polymorphism associated with risk of neuromyelitis optica. Neurobiol. Dis. 2010, 37, 349–355. [Google Scholar] [CrossRef]
- Zhao, G.X.; Liu, Y.; Li, Z.X.; Lv, C.Z.; Traboulsee, A.; Sadovnick, A.D.; Wu, Z.Y. Variants in the promoter region of CYP7A1 are associated with neuromyelitis optica but not with multiple sclerosis in the Han Chinese population. Neurosci. Bull. 2013, 29, 525–530. [Google Scholar] [CrossRef]
- Xu, Y.; Li, L.; Ren, H.T.; Yin, B.; Yuan, J.G.; Peng, X.Z.; Qiang, B.Q.; Cui, L.Y. Mutation of the cellular adhesion molecule NECL2 is associated with neuromyelitis optica spectrum disorder. J. Neurol. Sci. 2018, 388, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Yanyu, C.; Rui, L.; Caixia, L.; Youming, L.; Jianhua, H.; Weihua, M.; Xiaobo, S.; Wen, X.; Ying, C.; et al. Human aquaporin 4 gene polymorphisms in Chinese patients with neuromyelitis optica. J. Neuroimmunol. 2014, 274, 192–196. [Google Scholar] [CrossRef]
- Lan, W.; Fang, S.; Zhang, H.; Wang, D.T.J.; Wu, J. The Fc Receptor-Like 3 Polymorphisms (rs7528684, rs945635, rs3761959 and rs2282284) and The Risk of Neuromyelitis Optica in A Chinese Population. Medicine (Baltimore) 2015, 94, e1320. [Google Scholar] [CrossRef]
- Shin, J.G.; Kim, H.J.; Park, B.L.; Bae, J.S.; Kim, L.H.; Cheong, H.S.; Shin, H.D. Putative association of GPC5 polymorphism with the risk of inflammatory demyelinating diseases. J. Neurol. Sci. 2013, 335, 82–88. [Google Scholar] [CrossRef]
- Matsushita, T.; Masaki, K.; Isobe, N.; Sato, S.; Yamamoto, K.; Nakamura, Y.; Watanabe, M.; Suenaga, T.; Kira, J.I.; Japan Multiple Sclerosis Genetic, C. Genetic factors for susceptibility to and manifestations of neuromyelitis optica. Ann. Clin. Transl. Neurol. 2020, 7, 2082–2093. [Google Scholar] [CrossRef]
- Mei, S.; Li, X.; Gong, X.; Li, X.; Yang, L.; Zhou, H.; Liu, Y.; Zhou, A.; Zhu, L.; Zhang, X.; et al. LC-MS/MS Analysis of Erythrocyte Thiopurine Nucleotides and Their Association with Genetic Variants in Patients With Neuromyelitis Optica Spectrum Disorders Taking Azathioprine. Ther. Drug. Monit. 2017, 39, 5–12. [Google Scholar] [CrossRef]
- Dai, Y.; Li, J.; Zhong, X.; Wang, Y.; Qiu, W.; Lu, Z.; Wu, A.; Bao, J.; Peng, F.; Hu, X. IL2RA Allele Increases Risk of Neuromyelitis Optica in Southern Han Chinese. Can. J. Neurol. Sci. 2013, 40, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Bae, J.S.; Kim, H.J.; Shin, H.D. CD58 polymorphisms associated with the risk of neuromyelitis optica in a Korean population. BMC Neurol. 2014, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Kim, H.J.; Kim, J.H.; Bae, J.S.; Cheong, H.S.; Park, B.L.; Shin, H.D. Associations of CD6, TNFRSF1A and IRF8 polymorphisms with risk of inflammatory demyelinating diseases. Neuropathol. Appl. Neurobiol. 2013, 39, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, Z.; Lian, Z.; Chen, H.; Zhang, Q.; Feng, H.; Miao, X.; Du, Q.; Zhou, H. Association of CD58 gene polymorphisms with NMO spectrum disorders in a Han Chinese population. J. Neuroimmunol. 2017, 309, 23–30. [Google Scholar] [CrossRef]
- Zhuang, J.C.; Wu, L.; Qian, M.Z.; Cai, P.P.; Liu, Q.B.; Zhao, G.X.; Li, Z.X.; Wu, Z.Y. Variants of Interleukin-7/Interleukin-7 Receptor Alpha are Associated with Both Neuromyelitis Optica and Multiple Sclerosis Among Chinese Han Population in Southeastern China. Chin. Med. J. (Engl.) 2015, 128, 3062–3068. [Google Scholar] [CrossRef]
- Liu, C.; Wang, G.; Liu, H.; Li, Y.; Li, J.; Dai, Y.; Hu, X. CD226 Gly307Ser association with neuromyelitis optica in Southern Han Chinese. Can. J. Neurol. Sci. 2012, 39, 488–490. [Google Scholar] [CrossRef]
- Bruijstens, A.L.; Wong, Y.Y.M.; van Pelt, D.E.; van der Linden, P.J.E.; Haasnoot, G.W.; Hintzen, R.Q.; Claas, F.H.J.; Neuteboom, R.F.; Wokke, B.H.A. HLA association in MOG-IgG- and AQP4-IgG-related disorders of the CNS in the Dutch population. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e702. [Google Scholar] [CrossRef]
- Kay, C.S.K.; Scola, R.H.; Arndt, R.C.; Lorenzoni, P.J.; Werneck, L.C. HLA-alleles class I and II associated with genetic susceptibility to neuromyelitis optica in Brazilian patients. Arq. Neuropsiquiatr. 2019, 77, 239–247. [Google Scholar] [CrossRef]
- Alvarenga, M.P.; Fernandez, O.; Leyva, L.; Campanella, L.; Vasconcelos, C.F.; Alvarenga, M.; Papais Alvarenga, R.M. The HLA DRB1*03:01 allele is associated with NMO regardless of the NMO-IgG status in Brazilian patients from Rio de Janeiro. J. Neuroimmunol. 2017, 310, 1–7. [Google Scholar] [CrossRef]
- Deschamps, R.; Paturel, L.; Jeannin, S.; Chausson, N.; Olindo, S.; Bera, O.; Bellance, R.; Smadja, D.; Cesaire, D.; Cabre, P. Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro-Caribbean population. Mult. Scler. 2011, 17, 24–31. [Google Scholar] [CrossRef]
- Romero-Hidalgo, S.; Flores-Rivera, J.; Rivas-Alonso, V.; Barquera, R.; Villarreal-Molina, M.T.; Antuna-Puente, B.; Macias-Kauffer, L.R.; Villalobos-Comparan, M.; Ortiz-Maldonado, J.; Yu, N.; et al. Native American ancestry significantly contributes to neuromyelitis optica susceptibility in the admixed Mexican population. Sci. Rep. 2020, 10, 13706. [Google Scholar] [CrossRef] [PubMed]
- Zephir, H.; Fajardy, I.; Outteryck, O.; Blanc, F.; Roger, N.; Fleury, M.; Rudolf, G.; Marignier, R.; Vukusic, S.; Confavreux, C.; et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult. Scler. 2009, 15, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Okuno, T.; Hosomichi, K.; Hosokawa, A.; Hirata, J.; Suzuki, K.; Sakaue, S.; Kinoshita, M.; Asano, Y.; Miyamoto, K.; et al. Next-generation sequencing identifies contribution of both class I and II HLA genes on susceptibility of multiple sclerosis in Japanese. J. Neuroinflammation 2019, 16, 162. [Google Scholar] [CrossRef]
- Hofer, L.S.; Ramberger, M.; Gredler, V.; Pescoller, A.S.; Rostasy, K.; Sospedra, M.; Hegen, H.; Berger, T.; Lutterotti, A.; Reindl, M. Comparative Analysis of T-Cell Responses to Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein in Inflammatory Demyelinating Central Nervous System Diseases. Front. Immunol. 2020, 11, 1188. [Google Scholar] [CrossRef] [PubMed]
- Estrada, K.; Whelan, C.W.; Zhao, F.; Bronson, P.; Handsaker, R.E.; Sun, C.; Carulli, J.P.; Harris, T.; Ransohoff, R.M.; McCarroll, S.A.; et al. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat. Commun. 2018, 9, 1929. [Google Scholar] [CrossRef]
- Arnold, A.C. Evolving management of optic neuritis and multiple sclerosis. Am. J. Ophthalmol. 2005, 139, 1101–1108. [Google Scholar] [CrossRef]
- Chen, L.; Gordon, L.K. Ocular manifestations of multiple sclerosis. Curr. Opin. Ophthalmol. 2005, 16, 315–320. [Google Scholar] [CrossRef]
- De Santi, L.; Annunziata, P. Symptomatic cranial neuralgias in multiple sclerosis: Clinical features and treatment. Clin. Neurol. Neurosurg. 2012, 114, 101–107. [Google Scholar] [CrossRef]
- Rizzo, J.F., 3rd; Lessell, S. Risk of developing multiple sclerosis after uncomplicated optic neuritis: A long-term prospective study. Neurology 1988, 38, 185–190. [Google Scholar] [CrossRef]
- Francis, D.A.; Compston, D.A.; Batchelor, J.R.; McDonald, W.I. A reassessment of the risk of multiple sclerosis developing in patients with optic neuritis after extended follow-up. J. Neurol. Neurosurg. Psychiatry 1987, 50, 758–765. [Google Scholar] [CrossRef]
- Dalton, C.M.; Brex, P.A.; Miszkiel, K.A.; Fernando, K.; MacManus, D.G.; Plant, G.T.; Thompson, A.J.; Miller, D.H. Spinal cord MRI in clinically isolated optic neuritis. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Zimprich, A.; Martinelli-Boneschi, F.; Herculano, B.; Wang, Z.; Matesanz, F.; Urcelay, E.; Vandenbroeck, K.; Leyva, L.; Gris, D.; et al. Exome sequencing in multiple sclerosis families identifies 12 candidate genes and nominates biological pathways for the genesis of disease. PLoS Genet. 2019, 15, e1008180. [Google Scholar] [CrossRef] [PubMed]
- Gil-Varea, E.; Urcelay, E.; Vilarino-Guell, C.; Costa, C.; Midaglia, L.; Matesanz, F.; Rodriguez-Antiguedad, A.; Oksenberg, J.; Espino-Paisan, L.; Dessa Sadovnick, A.; et al. Exome sequencing study in patients with multiple sclerosis reveals variants associated with disease course. J. Neuroinflammation 2018, 15, 265. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Morimoto, C.; Burks, J.S.; Kerr, C.; Hauser, S.L. Dual-label immunocytochemistry of the active multiple sclerosis lesion: Major histocompatibility complex and activation antigens. Ann. Neurol. 1988, 24, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Mews, I.; Bergmann, M.; Bunkowski, S.; Gullotta, F.; Bruck, W. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult. Scler. 1998, 4, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Roostaei, T.; Sadaghiani, S.; Mashhadi, R.; Falahatian, M.; Mohamadi, E.; Javadian, N.; Nazeri, A.; Doosti, R.; Naser Moghadasi, A.; Owji, M.; et al. Convergent effects of a functional C3 variant on brain atrophy, demyelination, and cognitive impairment in multiple sclerosis. Mult. Scler. 2019, 25, 532–540. [Google Scholar] [CrossRef]
- Tassoni, A.; Farkhondeh, V.; Itoh, Y.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep. 2019, 9, 10010. [Google Scholar] [CrossRef]
- Szalai, A.J.; Hu, X.; Adams, J.E.; Barnum, S.R. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol. Immunol. 2007, 44, 3132–3136. [Google Scholar] [CrossRef]
- Lipsker, D. The schnitzler syndrome. Orphanet J. Rare Dis. 2010, 5, 38. [Google Scholar] [CrossRef]
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet. Med. 2016, 18, 483–493. [Google Scholar] [CrossRef]
- Lewis, J.M. COCKAYNE SYNDROME (CS) MASQUERADING AS SECKEL SYNDROME (SS). Pediatric Res. 1987, 21, 229. [Google Scholar] [CrossRef][Green Version]
- Wierzbicki, A.S.; Lloyd, M.D.; Schofield, C.J.; Feher, M.D.; Gibberd, F.B. Refsum’s disease: A peroxisomal disorder affecting phytanic acid alpha-oxidation. J. Neurochem. 2002, 80, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Ji, H.; Yan, H.; Wang, J.; Zhang, Y.; Zhang, Q.; Li, D.; Cao, B.; Gu, Q.; Wu, Y.; et al. Genotype-phenotype correlation and natural history analyses in a Chinese cohort with pelizaeus-merzbacher disease. Orphanet J. Rare Dis. 2022, 17, 137. [Google Scholar] [CrossRef] [PubMed]
- Maria, B.L.; Quisling, R.G.; Rosainz, L.C.; Yachnis, A.T.; Gitten, J.; Dede, D.; Fennell, E. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J. Child Neurol. 1999, 14, 368–376. [Google Scholar] [CrossRef]
- Maria, B.L.; Hoang, K.B.; Tusa, R.J.; Mancuso, A.A.; Hamed, L.M.; Quisling, R.G.; Hove, M.T.; Fennell, E.B.; Booth-Jones, M.; Ringdahl, D.M.; et al. “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997, 12, 423–430. [Google Scholar] [CrossRef]
- Parisi, M.A.; Doherty, D.; Chance, P.F.; Glass, I.A. Joubert syndrome (and related disorders) (OMIM 213300). Eur. J. Hum. Genet. 2007, 15, 511–521. [Google Scholar] [CrossRef]
- Maria, B.L.; Bozorgmanesh, A.; Kimmel, K.N.; Theriaque, D.; Quisling, R.G. Quantitative assessment of brainstem development in Joubert syndrome and Dandy-Walker syndrome. J. Child. Neurol. 2001, 16, 751–758. [Google Scholar] [CrossRef]
- Doherty, D. Joubert syndrome: Insights into brain development, cilium biology, and complex disease. Semin. Pediatr. Neurol. 2009, 16, 143–154. [Google Scholar] [CrossRef]
- Wang, S.F.; Kowal, T.J.; Ning, K.; Koo, E.B.; Wu, A.Y.; Mahajan, V.B.; Sun, Y. Review of Ocular Manifestations of Joubert Syndrome. Genes 2018, 9, 605. [Google Scholar] [CrossRef]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome-Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.N.; Bhan, A.; Heon, E. Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet-Biedl syndrome. Clin. Genet. 2016, 89, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Katsanis, N. Bardet-Biedl Syndrome. In Polycystic Kidney Disease: Translating Mechanisms into Therapy; Cowley, J.B.D., Bissler, J.J., Eds.; Springer: New York, NY, USA, 2018; pp. 27–50. [Google Scholar]
- Joy, T.; Cao, H.; Black, G.; Malik, R.; Charlton-Menys, V.; Hegele, R.A.; Durrington, P.N. Alstrom syndrome (OMIM 203800): A case report and literature review. Orphanet J. Rare Dis. 2007, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.D.; Beck, S.; Maffei, P.; Naggert, J.K. Alström syndrome. Eur. J. Hum. Genet. 2007, 15, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Raz, E.; Loh, J.P.; Saba, L.; Omari, M.; Herbert, J.; Lui, Y.; Kister, I. Periventricular lesions help differentiate neuromyelitis optica spectrum disorders from multiple sclerosis. Mult. Scler. Int. 2014, 2014, 986923. [Google Scholar] [CrossRef] [PubMed]
- Borazanci, A.P.; Harris, M.K.; Schwendimann, R.N.; Gonzalez-Toledo, E.; Maghzi, A.H.; Etemadifar, M.; Alekseeva, N.; Pinkston, J.; Kelley, R.E.; Minagar, A. Multiple sclerosis: Clinical features, pathophysiology, neuroimaging and future therapies. Future Neurol. 2009, 4, 229–246. [Google Scholar] [CrossRef]
- Mills, R.J.; Young, C.A.; Smith, E.T. Central trigeminal involvement in multiple sclerosis using high-resolution MRI at 3 T. Br. J. Radiol. 2010, 83, 493–498. [Google Scholar] [CrossRef]
- Ghezzi, A.; Deplano, V.; Faroni, J.; Grasso, M.G.; Liguori, M.; Marrosu, G.; Pozzilli, C.; Simone, I.L.; Zaffaroni, M. Multiple sclerosis in childhood: Clinical features of 149 cases. Mult. Scler. 1997, 3, 43–46. [Google Scholar] [CrossRef]
- Putzki, N.; Pfriem, A.; Limmroth, V.; Yaldizli, O.; Tettenborn, B.; Diener, H.C.; Katsarava, Z. Prevalence of migraine, tension-type headache and trigeminal neuralgia in multiple sclerosis. Eur. J. Neurol. 2009, 16, 262–267. [Google Scholar] [CrossRef]
- van Dijkman, S.C.; de Jager, N.C.B.; Rauwé, W.M.; Danhof, M.; Della Pasqua, O. Effect of Age-Related Factors on the Pharmacokinetics of Lamotrigine and Potential Implications for Maintenance Dose Optimisation in Future Clinical Trials. Clin. Pharm. 2018, 57, 1039–1053. [Google Scholar] [CrossRef]
- Cruccu, G.; Biasiotta, A.; Di Rezze, S.; Fiorelli, M.; Galeotti, F.; Innocenti, P.; Mameli, S.; Millefiorini, E.; Truini, A. Trigeminal neuralgia and pain related to multiple sclerosis. Pain 2009, 143, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.I.; Bronstein, A.M.; Gresty, M.A.; Du Boulay, E.P.; Rudge, P. Clinical and MRI correlates in 27 patients with acquired pendular nystagmus. Brain 1996, 119 Pt 2, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sasayama, H.; Mizuta, I.; Okamoto, Y.; Yoshida, M.; Riku, Y.; Hayashi, Y.; Yonezu, T.; Takata, Y.; Ohnari, K.; et al. Glial fibrillary acidic protein mutations in adult-onset Alexander disease: Clinical features observed in 12 Japanese patients. Acta Neurol. Scand. 2011, 124, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T. Clinical characteristics of Alexander disease. Neurodegener. Dis. Manag. 2020, 10, 325–333. [Google Scholar] [CrossRef]
- Balbi, P.; Salvini, S.; Fundarò, C.; Frazzitta, G.; Maestri, R.; Mosah, D.; Uggetti, C.; Sechi, G. The clinical spectrum of late-onset Alexander disease: A systematic literature review. J. Neurol. 2010, 257, 1955–1962. [Google Scholar] [CrossRef]
- Yoshida, T.; Mizuta, I.; Yasuda, R.; Nakagawa, M.; Mizuno, T. Characteristics of cerebral lesions in adult-onset Alexander disease. Neurol. Sci. 2020, 41, 225–227. [Google Scholar] [CrossRef]
- Adam, M.P.; Schelley, S.; Gallagher, R.; Brady, A.N.; Barr, K.; Blumberg, B.; Shieh, J.T.; Graham, J.; Slavotinek, A.; Martin, M.; et al. Clinical features and management issues in Mowat-Wilson syndrome. Am. J. Med. Genet. A 2006, 140, 2730–2741. [Google Scholar] [CrossRef]
- Horn, D.; Weschke, B.; Zweier, C.; Rauch, A. Facial phenotype allows diagnosis of Mowat-Wilson syndrome in the absence of Hirschsprung disease. Am. J. Med. Genet. A 2004, 124, 102–104. [Google Scholar] [CrossRef]
- Hoffer, M.J.; Hilhorst-Hofstee, Y.; Knijnenburg, J.; Hansson, K.B.; Engelberts, A.C.; Laan, L.A.; Bakker, E.; Rosenberg, C. A 6Mb deletion in band 2q22 due to a complex chromosome rearrangement associated with severe psychomotor retardation, microcephaly and distinctive dysmorphic facial features. Eur. J. Med. Genet. 2007, 50, 149–154. [Google Scholar] [CrossRef]
- Silengo, M.; Ferrero, G.B.; Tornetta, L.; Cortese, M.G.; Canavese, F.; D’Alonzo, G.; Papalia, F. Pachygyria and cerebellar hypoplasia in Goldberg-Shprintzen syndrome. Am. J. Med. Genet. A 2003, 118, 388–390. [Google Scholar] [CrossRef]
- Garavelli, L.; Ivanovski, I.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; Baldo, C.; et al. Neuroimaging findings in Mowat-Wilson syndrome: A study of 54 patients. Genet. Med. 2017, 19, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Zweier, C.; Thiel, C.T.; Dufke, A.; Crow, Y.J.; Meinecke, P.; Suri, M.; Ala-Mello, S.; Beemer, F.; Bernasconi, S.; Bianchi, P.; et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur. J. Med. Genet. 2005, 48, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Christodoulou, J. MECP2 Disorders. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Han, Z.A.; Jeon, H.R.; Kim, S.W.; Park, J.Y.; Chung, H.J. Clinical characteristics of children with rett syndrome. Ann. Rehabil. Med. 2012, 36, 334–339. [Google Scholar] [CrossRef]
- de Breet, L.H.M.; Townend, G.S.; Curfs, L.M.G.; Kingma, H.; Smeets, E.E.J.; Lucieer, F.; Widdershoven, J.; van de Berg, R. Challenges in evaluating the oculomotor function in individuals with Rett syndrome using electronystagmography. Eur. J. Paediatr. Neurol. 2019, 23, 262–269. [Google Scholar] [CrossRef]
- Rose, S.A.; Djukic, A.; Jankowski, J.J.; Feldman, J.F.; Fishman, I.; Valicenti-McDermott, M. Rett syndrome: An eye-tracking study of attention and recognition memory. Dev. Med. Child Neurol. 2013, 55, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.A.; Wass, S.; Jankowski, J.J.; Feldman, J.F.; Djukic, A. Impaired Visual Search in Children with Rett Syndrome. Pediatr. Neurol. 2019, 92, 26–31. [Google Scholar] [CrossRef]
- Ji, H.; Huang, Z.; Xia, Z.; Molokeev, M.S.; Jiang, X.; Lin, Z.; Atuchin, V.V. Comparative investigations of the crystal structure and photoluminescence property of eulytite-type Ba3Eu(PO4)3 and Sr3Eu(PO4)3. Dalton Trans. 2015, 44, 7679–7686. [Google Scholar] [CrossRef]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne Syndrome. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Stevenson, R.E. Alpha-Thalassemia X-Linked Intellectual Disability Syndrome. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Gibbons, R. Alpha thalassaemia-mental retardation, X linked. Orphanet J. Rare Dis. 2006, 1, 15. [Google Scholar] [CrossRef]
- Medina, C.F.; Mazerolle, C.; Wang, Y.; Berube, N.G.; Coupland, S.; Gibbons, R.J.; Wallace, V.A.; Picketts, D.J. Altered visual function and interneuron survival in Atrx knockout mice: Inference for the human syndrome. Hum. Mol. Genet. 2009, 18, 966–977. [Google Scholar] [CrossRef]
- Wang, K.Y.; Chetta, J.; Bains, P.; Balzer, A.; Lincoln, J.; Uribe, T.; Lincoln, C.M. Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder: A pictorial review. Br. J. Radiol. 2018, 91, 20170690. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Dong, H.; Lee, H.; Liu, Z.; Tong, Y.; Elkin, K.; Haddad, Y.; Geng, X.; Ding, Y. Clinical Characteristics and Outcomes of Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder With Brainstem Lesions as Heralding Prodrome. Front. Neurol. 2022, 13, 836337. [Google Scholar] [CrossRef] [PubMed]
- Ashtari, F.; Safaei, A.; Shaygannejad, V.; Najafi, M.A.; Vesal, S. Neuromyelitis optica spectrum disease characteristics in Isfahan, Iran: A cross-sectional study. J. Res. Med. Sci. 2017, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.G.; Silva, I.T.F.; Dos Santos, T.S.S.; Filho, M.B.P.; de Abreu, F.F.; Oliveira-Filho, J. Clinical and prognostic aspects of patients with the Neuromyelitis Optica Spectrum Disorder (NMOSD) from a cohort in Northeast Brazil. BMC Neurol. 2022, 22, 95. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef]
- Huang, L.; Szymanska, K.; Jensen, V.L.; Janecke, A.R.; Innes, A.M.; Davis, E.E.; Frosk, P.; Li, C.; Willer, J.R.; Chodirker, B.N. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 2011, 89, 713–730. [Google Scholar] [CrossRef]
- Sheng, G.; Xu, X.; Lin, Y.F.; Wang, C.E.; Rong, J.; Cheng, D.; Peng, J.; Jiang, X.; Li, S.H.; Li, X.J. Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Investig. 2008, 118, 2785–2795. [Google Scholar] [CrossRef]
- Bennett, P.; Glass, I.; Swaid, S.; Dohayan, N.; Bakhsh, E.; Indridason, O.; Dobyns, W.; Parisi, C.; Doherty, D.; Eckert, M. mutations cause both retinal dystrophy and AHI1. J. Med. Genet. 2006, 43, 334–339. [Google Scholar] [CrossRef]
- Gazea, M.; Tasouri, E.; Heigl, T.; Bosch, V.; Tucker, K.L.; Blaess, S. Definition of a critical spatiotemporal window within which primary cilia control midbrain dopaminergic neurogenesis. Neurogenesis (Austin) 2016, 3, e1248206. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Gopal, D.J.; Kim, J.; Saleem, S.N.; Silhavy, J.L.; Louie, C.M.; Thacker, B.E.; Williams, Y.; Zaki, M.S.; Gleeson, J.G. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med. 2011, 17, 726–731. [Google Scholar] [CrossRef]
- Martemyanov, K.A. G protein signaling in the retina and beyond: The Cogan lecture. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8201–8207. [Google Scholar] [CrossRef] [PubMed]
- Rajala, R.V.S. Signaling roles of phosphoinositides in the retina. J. Lipid. Res. 2021, 62, 100041. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Berbari, N.F.; O’Connor, A.K.; Haycraft, C.J.; Yoder, B.K. The primary cilium as a complex signaling center. Curr. Biol. 2009, 19, R526-35. [Google Scholar] [CrossRef]
- Guo, J.; Otis, J.M.; Suciu, S.K.; Catalano, C.; Xing, L.; Constable, S.; Wachten, D.; Gupton, S.; Lee, J.; Lee, A.; et al. Primary Cilia Signaling Promotes Axonal Tract Development and Is Disrupted in Joubert Syndrome-Related Disorders Models. Dev. Cell 2019, 51, 759–774.e5. [Google Scholar] [CrossRef] [PubMed]
- Sharif, A.S.; Gerstner, C.D.; Cady, M.A.; Arshavsky, V.Y.; Mitchell, C.; Ying, G.; Frederick, J.M.; Baehr, W. Deletion of the phosphatase INPP5E in the murine retina impairs photoreceptor axoneme formation and prevents disc morphogenesis. J. Biol. Chem. 2021, 296, 100529. [Google Scholar] [CrossRef]
- Vilboux, T.; Malicdan, M.C.; Roney, J.C.; Cullinane, A.R.; Stephen, J.; Yildirimli, D.; Bryant, J.; Fischer, R.; Vemulapalli, M.; Mullikin, J.C.; et al. CELSR2, encoding a planar cell polarity protein, is a putative gene in Joubert syndrome with cortical heterotopia, microophthalmia, and growth hormone deficiency. Am. J. Med. Genet. A 2017, 173, 661–666. [Google Scholar] [CrossRef]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nurnberg, P.; Nurnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). Elife 2015, 4, e08077. [Google Scholar] [CrossRef]
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mockel, A.; Perdomo, Y.; Stutzmann, F.; Letsch, J.; Marion, V.; Dollfus, H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011, 30, 258–274. [Google Scholar] [CrossRef]
- Kim, J.C.; Ou, Y.Y.; Badano, J.L.; Esmail, M.A.; Leitch, C.C.; Fiedrich, E.; Beales, P.L.; Archibald, J.M.; Katsanis, N.; Rattner, J.B.; et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J. Cell Sci. 2005, 118 Pt 5, 1007–1020. [Google Scholar] [CrossRef]
- Marion, V.; Stoetzel, C.; Schlicht, D.; Messaddeq, N.; Koch, M.; Flori, E.; Danse, J.M.; Mandel, J.L.; Dollfus, H. Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, D.; Seo, S.; Stone, E.M.; Sheffield, V.C. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J. Biol. Chem. 2012, 287, 20625–20635. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Baye, L.M.; Schulz, N.P.; Beck, J.S.; Zhang, Q.; Slusarski, D.C.; Sheffield, V.C. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Northam, G.B.; Chong, W.K.; Banks, T.; Beales, P.; Baldeweg, T. Neocortical and hippocampal volume loss in a human ciliopathy: A quantitative MRI study in Bardet-Biedl syndrome. Am. J. Med. Genet. A 2011, 155A, 1–8. [Google Scholar] [CrossRef]
- Hulleman, J.D.; Nguyen, A.; Ramprasad, V.L.; Murugan, S.; Gupta, R.; Mahindrakar, A.; Angara, R.; Sankurathri, C.; Mootha, V.V. A novel H395R mutation in MKKS/BBS6 causes retinitis pigmentosa and polydactyly without other findings of Bardet-Biedl or McKusick-Kaufman syndrome. Mol. Vis. 2016, 22, 73–81. [Google Scholar] [PubMed]
- FDA Approves Treatment for Weight Management in Patients with Bardet-Biedl Syndrome Aged 6 or Older. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-weight-management-patients-bardet-biedl-syndrome-aged-6-or-older (accessed on 31 July 2022).
- Prado, D.A.; Acosta-Acero, M.; Maldonado, R.S. Gene therapy beyond luxturna: A new horizon of the treatment for inherited retinal disease. Curr. Opin. Ophthalmol. 2020, 31, 147–154. [Google Scholar] [CrossRef]
- Seo, S.; Mullins, R.F.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Wang, K.; Stone, E.M.; Sheffield, V.; Drack, A.V. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6118–6132. [Google Scholar] [CrossRef]
- Knorz, V.J.; Spalluto, C.; Lessard, M.; Purvis, T.L.; Adigun, F.F.; Collin, G.B.; Hanley, N.A.; Wilson, D.I.; Hearn, T. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol. Biol. Cell 2010, 21, 3617–3629. [Google Scholar] [CrossRef]
- Alvarez-Satta, M.; Lago-Docampo, M.; Bea-Mascato, B.; Solarat, C.; Castro-Sanchez, S.; Christensen, S.T.; Valverde, D. ALMS1 Regulates TGF-beta Signaling and Morphology of Primary Cilia. Front. Cell Dev. Biol. 2021, 9, 623829. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Yang, J.; Adamian, M.; Li, T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 2006, 17, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015, 6, 7857. [Google Scholar] [CrossRef]
- Heydet, D.; Chen, L.X.; Larter, C.Z.; Inglis, C.; Silverman, M.A.; Farrell, G.C.; Leroux, M.R. A truncating mutation of Alms1 reduces the number of hypothalamic neuronal cilia in obese mice. Dev. Neurobiol. 2013, 73, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Messing, A.; Brenner, M.; Feany, M.B.; Nedergaard, M.; Goldman, J.E. Alexander disease. J. Neurosci. 2012, 32, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef]
- Gispert, J.D.; Monte, G.C.; Falcon, C.; Tucholka, A.; Rojas, S.; Sanchez-Valle, R.; Antonell, A.; Llado, A.; Rami, L.; Molinuevo, J.L. CSF YKL-40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol. Aging 2016, 38, 47–55. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Longo, A.; Lazzara, F.; Cambria, D.; Distefano, G.; Palumbo, M.; Cantarella, A.; Malaguarnera, L.; Di Rosa, M. CHI3L1 and CHI3L2 overexpression in motor cortex and spinal cord of sALS patients. Mol. Cell Neurosci. 2017, 85, 162–169. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wang, G.; Starkey, A.; Hamilton, R.L.; Wiley, C.A. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflammation. 2010, 7, 34. [Google Scholar] [CrossRef]
- Hinsinger, G.; Galeotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehmann, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Burman, J.; Raininko, R.; Blennow, K.; Zetterberg, H.; Axelsson, M.; Malmestrom, C. YKL-40 is a CSF biomarker of intrathecal inflammation in secondary progressive multiple sclerosis. J. Neuroimmunol. 2016, 292, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Unger, T.; Lewis, D.A.; Levitt, P.; Mirnics, K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol. Psychiatry 2007, 62, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.; Lou, H.; Zhang, J.; Tian, H.; Ou, Q.; Xu, J.Y.; Jin, C.; Gao, F.; Zhang, J.; Wang, J.; et al. MicroRNA-24 protects retina from degeneration in rats by down-regulating chitinase-3-like protein 1. Exp. Eye Res. 2019, 188, 107791. [Google Scholar] [CrossRef] [PubMed]
- Fraher, J.P.; Dockery, P.; O’Donoghue, O.; Riedewald, B.; O’Leary, D. Initial motor axon outgrowth from the developing central nervous system. J. Anat. 2007, 211, 600–611. [Google Scholar] [CrossRef]
- Nazareth, L.; Chen, M.; Shelper, T.; Shah, M.; Tello Velasquez, J.; Walkden, H.; Beacham, I.; Batzloff, M.; Rayfield, A.; Todorovic, M.; et al. Novel insights into the glia limitans of the olfactory nervous system. J. Comp. Neurol. 2019, 527, 1228–1244. [Google Scholar] [CrossRef]
- Monzon-Mayor, M.; Yanes, C.; Ghandour, M.S.; de Barry, J.; Gombos, G. Glial fibrillary acidic protein and vimentin immunohistochemistry in the developing and adult midbrain of the lizard Gallotia galloti. J. Comp. Neurol. 1990, 295, 569–579. [Google Scholar] [CrossRef]
- Gow, A.; Sharma, R. The unfolded protein response in protein aggregating diseases. Neuromolecular Med. 2003, 4, 73–94. [Google Scholar] [CrossRef]
- Garbern, J.Y. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell. Mol. Life Sci. 2007, 64, 50–65. [Google Scholar] [CrossRef]
- Southwood, C.M.; Garbern, J.; Jiang, W.; Gow, A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 2002, 36, 585–596. [Google Scholar] [CrossRef]
- Sistermans, E.A.; de Coo, R.F.; De Wijs, I.J.; Van Oost, B.A. Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology 1998, 50, 1749–1754. [Google Scholar] [CrossRef]
- Nobuta, H.; Yang, N.; Ng, Y.H.; Marro, S.G.; Sabeur, K.; Chavali, M.; Stockley, J.H.; Killilea, D.W.; Walter, P.B.; Zhao, C.; et al. Oligodendrocyte Death in Pelizaeus-Merzbacher Disease Is Rescued by Iron Chelation. Cell Stem Cell 2019, 25, 531–541.e6. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.L.; Powers, B.; Mazur, C.; Kim, A.; Wheeler, S.; Hung, G.; Swayze, E.; Messing, A. Antisense suppression of glial fibrillary acidic protein as a treatment for Alexander disease. Ann. Neurol. 2018, 83, 27–39. [Google Scholar] [CrossRef]
- Hippert, C.; Graca, A.B.; Basche, M.; Kalargyrou, A.A.; Georgiadis, A.; Ribeiro, J.; Matsuyama, A.; Aghaizu, N.; Bainbridge, J.W.; Smith, A.J.; et al. RNAi-mediated suppression of vimentin or glial fibrillary acidic protein prevents the establishment of Muller glial cell hypertrophy in progressive retinal degeneration. Glia 2021, 69, 2272–2290. [Google Scholar] [CrossRef] [PubMed]
- Bargagna-Mohan, P.; Paranthan, R.R.; Hamza, A.; Dimova, N.; Trucchi, B.; Srinivasan, C.; Elliott, G.I.; Zhan, C.G.; Lau, D.L.; Zhu, H.; et al. Withaferin A targets intermediate filaments glial fibrillary acidic protein and vimentin in a model of retinal gliosis. J. Biol. Chem. 2010, 285, 7657–7669. [Google Scholar] [CrossRef] [PubMed]
- Elitt, M.S.; Barbar, L.; Shick, H.E.; Powers, B.E.; Maeno-Hikichi, Y.; Madhavan, M.; Allan, K.C.; Nawash, B.S.; Gevorgyan, A.S.; Hung, S.; et al. Suppression of proteolipid protein rescues Pelizaeus-Merzbacher disease. Nature 2020, 585, 397–403. [Google Scholar] [CrossRef]
- Crunkhorn, S. ASO rescues Pelizaeus-Merzbacher disease. Nat. Rev. Drug. Discov. 2020, 19, 512. [Google Scholar] [CrossRef] [PubMed]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5’-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Souopgui, J.; Van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet. 2008, 17, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; Van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Magloire, V.; Mercier, M.S.; Kullmann, D.M.; Pavlov, I. GABAergic Interneurons in Seizures: Investigating Causality with Optogenetics. Neuroscientist 2019, 25, 344–358. [Google Scholar] [CrossRef]
- Marın, O.; Anderson, S.A.; Rubenstein, J.L. Origin and molecular specification of striatal interneurons. J. Neurosci. 2000, 20, 6063–6076. [Google Scholar] [CrossRef]
- McKinsey, G.L.; Lindtner, S.; Trzcinski, B.; Visel, A.; Pennacchio, L.A.; Huylebroeck, D.; Higashi, Y.; Rubenstein, J.L. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013, 77, 83–98. [Google Scholar] [CrossRef]
- Srivatsa, S.; Parthasarathy, S.; Molnar, Z.; Tarabykin, V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 2015, 85, 998–1012. [Google Scholar] [CrossRef]
- Fame, R.M.; MacDonald, J.L.; Macklis, J.D. Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 2011, 34, 41–50. [Google Scholar] [CrossRef]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef]
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Seuntjens, E.; Nityanandam, A.; Miquelajauregui, A.; Debruyn, J.; Stryjewska, A.; Goebbels, S.; Nave, K.A.; Huylebroeck, D.; Tarabykin, V. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat. Neurosci. 2009, 12, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Parrilla, Y.; Amiel, J.; Augé, J.; Encha-Razavi, F.; Munnich, A.; Lyonnet, S.; Vekemans, M.; Attié-Bitach, T. Expression of the SMADIP1 gene during early human development. Mech. Dev. 2002, 114, 187–191. [Google Scholar] [CrossRef]
- Bassez, G.; Camand, O.J.; Cacheux, V.; Kobetz, A.; Dastot-Le Moal, F.; Marchant, D.; Catala, M.; Abitbol, M.; Goossens, M. Pleiotropic and diverse expression of ZFHX1B gene transcripts during mouse and human development supports the various clinical manifestations of the “Mowat-Wilson” syndrome. Neurobiol. Dis. 2004, 15, 240–250. [Google Scholar] [CrossRef]
- Ariss, M.; Natan, K.; Friedman, N.; Traboulsi, E.I. Ophthalmologic abnormalities in Mowat-Wilson syndrome and a mutation in ZEB2. Ophthalmic Genet. 2012, 33, 159–160. [Google Scholar] [CrossRef]
- Forrest, M.P.; Waite, A.J.; Martin-Rendon, E.; Blake, D.J. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS ONE 2013, 8, e73169. [Google Scholar] [CrossRef]
- Flora, A.; Garcia, J.J.; Thaller, C.; Zoghbi, H.Y. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 15382–15387. [Google Scholar] [CrossRef]
- Malvaez, M.; Greenfield, V.Y.; Matheos, D.P.; Angelillis, N.A.; Murphy, M.D.; Kennedy, P.J.; Wood, M.A.; Wassum, K.M. Habits Are Negatively Regulated by Histone Deacetylase 3 in the Dorsal Striatum. Biol. Psychiatry 2018, 84, 383–392. [Google Scholar] [CrossRef]
- Townend, G.S.; van de Berg, R.; de Breet, L.H.M.; Hiemstra, M.; Wagter, L.; Smeets, E.; Widdershoven, J.; Kingma, H.; Curfs, L.M.G. Oculomotor Function in Individuals With Rett Syndrome. Pediatr. Neurol. 2018, 88, 48–58. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef]
- Hite, K.C.; Kalashnikova, A.A.; Hansen, J.C. Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Sci. 2012, 21, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Woodcock, C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, P.A.; Glass, C.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef]
- Carro, S.; Bergo, A.; Mengoni, M.; Bachi, A.; Badaracco, G.; Kilstrup-Nielsen, C.; Landsberger, N. A novel protein, Xenopus p20, influences the stability of MeCP2 through direct interaction. J. Biol. Chem. 2004, 279, 25623–25631. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef]
- Suzuki, M.; Yamada, T.; Kihara-Negishi, F.; Sakurai, T.; Oikawa, T. Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene 2003, 22, 8688–8698. [Google Scholar] [CrossRef]
- Buschdorf, J.P.; Stratling, W.H. A WW domain binding region in methyl-CpG-binding protein MeCP2: Impact on Rett syndrome. J. Mol. Med. (Berl.) 2004, 82, 135–143. [Google Scholar] [CrossRef]
- Harikrishnan, K.N.; Chow, M.Z.; Baker, E.K.; Pal, S.; Bassal, S.; Brasacchio, D.; Wang, L.; Craig, J.M.; Jones, P.L.; Sif, S.; et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 2005, 37, 254–264. [Google Scholar] [CrossRef]
- Jeffery, L.; Nakielny, S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 2004, 279, 49479–49487. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2019, Online ahead of print. [Google Scholar] [CrossRef]
- Connolly, D.R.; Zhou, Z. Genomic insights into MeCP2 function: A role for the maintenance of chromatin architecture. Curr. Opin. Neurobiol. 2019, 59, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Good, K.V.; Vincent, J.B.; Ausio, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Martinez de Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 63. [Google Scholar] [CrossRef]
- Sheikh, T.I.; de Paz, A.M.; Akhtar, S.; Ausio, J.; Vincent, J.B. MeCP2_E1 N-terminal modifications affect its degradation rate and are disrupted by the Ala2Val Rett mutation. Hum. Mol. Genet. 2017, 26, 4132–4141. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; de Lagarde, D.; Leonard, H.; Williamson, S.L.; Vasudevan, V.; Christodoulou, J.; Thompson, E.; MacLeod, P.; Ravine, D. Lost in translation: Translational interference from a recurrent mutation in exon 1 of MECP2. J. Med. Genet. 2006, 43, 470–477. [Google Scholar] [CrossRef][Green Version]
- Kharrat, M.; Hsairi, I.; Fendri-Kriaa, N.; Kenoun, H.; Othmen, H.B.; Ben Mahmoud, A.; Ghorbel, R.; Abid, I.; Triki, C.; Fakhfakh, F. A Novel Mutation p. A59P in N-Terminal Domain of Methyl-CpG–Binding Protein 2 Confers Phenotypic Variability in 3 Cases of Tunisian Rett Patients: Clinical Evaluations and In Silico Investigations. J. Child Neurol. 2015, 30, 1715–1721. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, J.; Zhang, Q.; Chen, Y.; Wu, X.; Bao, X. MECP2 mutation spectrum and its clinical characteristics in a Chinese cohort. Clin. Genet. 2020, 98, 240–250. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Yang, Y.; Uvarov, O.; Cao, W.; Alexov, E. Impact of Rett Syndrome Mutations on MeCP2 MBD Stability. Biochemistry 2015, 54, 6357–6368. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef]
- Ghosh, R.P.; Horowitz-Scherer, R.A.; Nikitina, T.; Gierasch, L.M.; Woodcock, C.L. Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J. Biol. Chem. 2008, 283, 20523–20534. [Google Scholar] [CrossRef] [PubMed]
- Spiga, O.; Gardini, S.; Rossi, N.; Cicaloni, V.; Pettini, F.; Niccolai, N.; Santucci, A. Structural investigation of Rett-inducing MeCP2 mutations. Genes Dis. 2019, 6, 31–34. [Google Scholar] [CrossRef] [PubMed]
- 344 Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Jellinger, K.; Seitelberger, F. Neuropathology of Rett syndrome. Am. J. Med. Genet. Suppl. 1986, 1, 259–288. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Microscopic observations of the brain in Rett syndrome. Neuropediatrics 1995, 26, 105–108. [Google Scholar] [CrossRef]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.; Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef]
- Kerr, A.M.; Stephenson, J.B. Rett’s syndrome in the west of Scotland. Br. Med. J. (Clin. Res. Ed.) 1985, 291, 579–582. [Google Scholar] [CrossRef]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, K.K.; Ross, P.D.; Hector, R.D.; Bahey, N.G.; Bailey, M.E.; Cobb, S.R. Gene therapy for Rett syndrome: Prospects and challenges. Future Neurol. 2015, 10, 467–484. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Brooks, A.I.; Stein, C.S.; Hughes, S.M.; Heth, J.; McCray, P.M., Jr.; Sauter, S.L.; Johnston, J.C.; Cory-Slechta, D.A.; Federoff, H.J.; Davidson, B.L. Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc. Natl. Acad. Sci. USA 2002, 99, 6216–6221. [Google Scholar] [CrossRef]
- Grieger, J.C.; Samulski, R.J. Adeno-associated virus as a gene therapy vector: Vector development, production and clinical applications. Gene Ther. Gene Deliv. Syst. 2005, 119–145. [Google Scholar] [CrossRef]
- Arruda, V.R.; Stedman, H.H.; Nichols, T.C.; Haskins, M.E.; Nicholson, M.; Herzog, R.W.; Couto, L.B.; High, K.A. Regional intravascular delivery of AAV-2-F. IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood 2005, 105, 3458–3464. [Google Scholar] [CrossRef]
- Gadalla, K.K.; Bailey, M.E.; Spike, R.C.; Ross, P.D.; Woodard, K.T.; Kalburgi, S.N.; Bachaboina, L.; Deng, J.V.; West, A.E.; Samulski, R.J.; et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol. Ther. 2013, 21, 18–30. [Google Scholar] [CrossRef]
- Na, E.S.; Nelson, E.D.; Adachi, M.; Autry, A.E.; Mahgoub, M.A.; Kavalali, E.T.; Monteggia, L.M. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 2012, 32, 3109–3117. [Google Scholar] [CrossRef]
- McGraw, C.M.; Samaco, R.C.; Zoghbi, H.Y. Adult neural function requires MeCP2. Science 2011, 333, 186. [Google Scholar] [CrossRef]
- Le, T.T.H.; Tran, N.T.; Dao, T.M.L.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kuhn, R.; Nguyen, T.L.; Rajewsky, K.; Chu, V.T. Efficient and Precise CRISPR/Cas9-Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front. Genet. 2019, 10, 625. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Lioy, D.T.; Cheval, H.; McGann, J.C.; Bissonnette, J.M.; Murtha, M.J.; Foust, K.D.; Kaspar, B.K.; Bird, A.; Mandel, G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 2013, 33, 13612–13620. [Google Scholar] [CrossRef] [PubMed]
- Matagne, V.; Ehinger, Y.; Saidi, L.; Borges-Correia, A.; Barkats, M.; Bartoli, M.; Villard, L.; Roux, J.C. A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol. Dis. 2017, 99, 1–11. [Google Scholar] [CrossRef]
- Gadalla, K.K.E.; Vudhironarit, T.; Hector, R.D.; Sinnett, S.; Bahey, N.G.; Bailey, M.E.S.; Gray, S.J.; Cobb, S.R. Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol. Ther. Methods Clin. Dev. 2017, 5, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Sinnett, S.E.; Hector, R.D.; Gadalla, K.K.E.; Heindel, C.; Chen, D.; Zaric, V.; Bailey, M.E.S.; Cobb, S.R.; Gray, S.J. Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Mol. Ther. Methods Clin. Dev. 2017, 5, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Sinnett, S.E.; Boyle, E.; Lyons, C.; Gray, S.J. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain 2021, 144, 3005–3019. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Min, Z.; Ji, Q.; Geng, L.; Su, Y.; Liu, Z.; Hu, H.; Wang, L.; Zhang, W.; Suzuiki, K.; et al. Rescue of premature aging defects in Cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell 2020, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy of CT103A Cells for Relapsed/Refractory Antibody-Associated Idiopathic Inflammatory Diseases of the Nervous System (CARTinNS). Available online: https://clinicaltrials.gov/ct2/show/NCT04561557 (accessed on 31 July 2022).
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and mutation independent therapy via CRISPR-Cas9 mediated cellular reprogramming in rod photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef]
- Li, P.; Kleinstiver, B.P.; Leon, M.Y.; Prew, M.S.; Navarro-Gomez, D.; Greenwald, S.H.; Pierce, E.A.; Joung, J.K.; Liu, Q. Allele-Specific CRISPR-Cas9 Genome Editing of the Single-Base P23H Mutation for Rhodopsin-Associated Dominant Retinitis Pigmentosa. Crispr. J. 2018, 1, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.T.; Wu, W.H.; Lee, T.T.; Wu, W.P.; Xu, C.L.; Park, K.S.; Cui, X.; Justus, S.; Lin, C.S.; Jauregui, R.; et al. Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa. Ophthalmology 2018, 125, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Cheng, T.; Yao, Y.; Li, X.; Ma, Y.; Li, L.; Zhao, H.; Bao, J.; Zhang, M.; Qiu, Z.; et al. In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 2019, 5, eaav3335. [Google Scholar] [CrossRef]
- Matson, S.W.; Bean, D.W.; George, J.W. DNA helicases: Enzymes with essential roles in all aspects of DNA metabolism. Bioessays 1994, 16, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Osenbroch, P.O.; Auk-Emblem, P.; Halsne, R.; Strand, J.; Forstrom, R.J.; van der Pluijm, I.; Eide, L. Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J. 2009, 276, 2811–2821. [Google Scholar] [CrossRef]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell. Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef]
- Wong, H.K.; Muftuoglu, M.; Beck, G.; Imam, S.Z.; Bohr, V.A.; Wilson, D.M. 3rd, Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007, 35, 4103–4113. [Google Scholar] [CrossRef]
- Iyama, T.; Lee, S.Y.; Berquist, B.R.; Gileadi, O.; Bohr, V.A.; Seidman, M.M.; McHugh, P.J.; Wilson, D.M. 3rd, CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015, 43, 247–258. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Fischer, E.S.; Scrima, A.; Bohm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.A.; Caputo, M.; Ye, T.; Donnio, L.M.; Proietti-de-Santis, L.; Coin, F.; Laugel, V.; Egly, J.M. Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol. Cell 2017, 68, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Rush, J.; Gygi, S.P.; Harper, J.W. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 2010, 143, 951–965. [Google Scholar] [CrossRef] [PubMed]
- Latini, P.; Frontini, M.; Caputo, M.; Gregan, J.; Cipak, L.; Filippi, S.; Kumar, V.; Velez-Cruz, R.; Stefanini, M.; Proietti-De-Santis, L. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle 2011, 10, 3719–3730. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Serrano, M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle 2010, 9, 4005–4012. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Rajagopal, G.; Levine, A.J. The tumor suppressor p53: Cancer and aging. Cell Cycle 2008, 7, 842–847. [Google Scholar] [CrossRef]
- Rapin, I.; Weidenheim, K.; Lindenbaum, Y.; Rosenbaum, P.; Merchant, S.N.; Krishna, S.; Dickson, D.W. Cockayne syndrome in adults: Review with clinical and pathologic study of a new case. J. Child Neurol. 2006, 21, 991–1006. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Tobias, E.S.; Tolmie, J.L.; Martin-Coignard, D.; Drouin-Garraud, V.; Valayannopoulos, V.; Sarasin, A.; Dollfus, H. Cerebro-oculo-facio-skeletal syndrome: Three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J. Med. Genet. 2008, 45, 564–571. [Google Scholar] [CrossRef]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; van Steeg, H.; Vijg, J. Aging and genome maintenance: Lessons from the mouse? Science 2003, 299, 1355–1359. [Google Scholar] [CrossRef]
- Yun, K.W.; Chae, S.A.; Lee, J.J.; Yun, S.W.; Yoo, B.H.; Lim, I.S.; Lee, M.K. The first case of X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome in Korea. J. Korean Med. Sci. 2011, 26, 146–149. [Google Scholar] [CrossRef]
- Hall, J.; Weksberg, R. Pediatric diseases and epigenetics. In Medical Epigenetics; Elsevier: Amsterdam, The Netherlands, 2021; pp. 377–406. [Google Scholar]
- Bowman, G.D.; Poirier, M.G. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Ivanauskiene, K.; Spirkoski, J.; Shah, A.; Vekterud, K.; Moskaug, J.O.; Boe, S.O.; Wong, L.H.; Kuntziger, T.; Collas, P. PML protein organizes heterochromatin domains where it regulates histone H3.3 deposition by ATRX/DAXX. Genome Res. 2017, 27, 913–921. [Google Scholar] [CrossRef]
- Berube, N.G.; Smeenk, C.A.; Picketts, D.J. Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum. Mol. Genet. 2000, 9, 539–547. [Google Scholar] [CrossRef]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Yang, J.C.; Law, M.J.; Amos, R.; Chapman, L.M.; Jelinska, C.; Garrick, D.; Clynes, D.; Gibbons, R.J.; Rhodes, D.; et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 2011, 18, 777–782. [Google Scholar] [CrossRef]
- Udugama, M.; FT, M.C.; Chan, F.L.; Tang, M.C.; Pickett, H.A.; JD, R.M.; Mayne, L.; Collas, P.; Mann, J.R.; Wong, L.H. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres. Nucleic Acids Res. 2015, 43, 10227–10237. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef]
- Cardoso, C.; Lutz, Y.; Mignon, C.; Compe, E.; Depetris, D.; Mattei, M.G.; Fontes, M.; Colleaux, L. ATR-X mutations cause impaired nuclear location and altered DNA binding properties of the XNP/ATR-X protein. J. Med. Genet. 2000, 37, 746–751. [Google Scholar] [CrossRef]
- Argentaro, A.; Yang, J.C.; Chapman, L.; Kowalczyk, M.S.; Gibbons, R.J.; Higgs, D.R.; Neuhaus, D.; Rhodes, D. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. USA 2007, 104, 11939–11944. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Wada, T.; Fisher, C.A.; Malik, N.; Mitson, M.J.; Steensma, D.P.; Fryer, A.; Goudie, D.R.; Krantz, I.D.; Traeger-Synodinos, J. Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 2008, 29, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Berube, N.G.; Healy, J.; Medina, C.F.; Wu, S.; Hodgson, T.; Jagla, M.; Picketts, D.J. Patient mutations alter ATRX targeting to PML nuclear bodies. Eur. J. Hum. Genet. 2008, 16, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- Watson, L.A.; Solomon, L.A.; Li, J.R.; Jiang, Y.; Edwards, M.; Shin-ya, K.; Beier, F.; Berube, N.G. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J. Clin. Investig. 2013, 123, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.; Seah, C.; Moulin, J.; Isaac, C.; Dick, F.; Berube, N.G. Loss of ATRX leads to chromosome cohesion and congression defects. J. Cell Biol. 2008, 180, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.; Watson, L.A.; Davidson, B.; Jiang, Y.; Berube, N.G. ATRX is required for maintenance of the neuroprogenitor cell pool in the embryonic mouse brain. Biol. Open 2014, 3, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef]
- Lagali, P.S.; Zhao, B.Y.H.; Yan, K.; Baker, A.N.; Coupland, S.G.; Tsilfidis, C.; Picketts, D.J. Sensory Experience Modulates Atrx-mediated Neuronal Integrity in the Mouse Retina. Neuroscience 2021, 452, 169–180. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Yamaguchi, K.; Onozato, M.; Li, Y.; Kurosawa, K.; Tanabe, H.; Okamoto, N.; Era, T.; Sugiyama, H.; et al. Targeting G-quadruplex DNA as cognitive function therapy for ATR-X syndrome. Nat. Med. 2018, 24, 802–813. [Google Scholar] [CrossRef]
- Wada, T.; Suzuki, S.; Shioda, N. 5-Aminolevulinic acid can ameliorate language dysfunction of patients with ATR-X syndrome. Congenit. Anom. (Kyoto) 2020, 60, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Poll-The, B.T.; Saudubray, J.M.; Ogier, H.A.; Odievre, M.; Scotto, J.M.; Monnens, L.; Govaerts, L.C.; Roels, F.; Cornelis, A.; Schutgens, R.B.; et al. Infantile Refsum disease: An inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy. Eur. J. Pediatr. 1987, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Datta, N.S.; Dobyns, W.B.; Hajra, A.K.; Moser, A.B.; Noetzel, M.J.; Zackai, E.H.; Moser, H.W. Neonatal adrenoleukodystrophy: New cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes. Am. J. Med. Genet. 1986, 23, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ebberink, M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta 2012, 1822, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Ciniawsky, S.; Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R.; Wendler, P. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat. Commun. 2015, 6, 7331. [Google Scholar] [CrossRef]
- Sugasini, D.; Yalagala, P.C.R.; Subbaiah, P.V. Efficient Enrichment of Retinal DHA with Dietary Lysophosphatidylcholine-DHA: Potential Application for Retinopathies. Nutrients 2020, 12, 3114. [Google Scholar] [CrossRef]
- Brites, P.; Mooyer, P.A.; El Mrabet, L.; Waterham, H.R.; Wanders, R.J. Plasmalogens participate in very-long-chain fatty acid-induced pathology. Brain 2009, 132 Pt 2, 482–492. [Google Scholar] [CrossRef]
- Verhoeven, N.M.; Jakobs, C. Human metabolism of phytanic acid and pristanic acid. Prog. Lipid Res. 2001, 40, 453–466. [Google Scholar] [CrossRef]
- Klouwer, F.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.; Engelen, M.; Poll-The, B.T. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare Dis. 2015, 10, 151. [Google Scholar] [CrossRef]
- Wills, A.J.; Manning, N.J.; Reilly, M.M. Refsum’s disease. QJM 2001, 94, 403–406. [Google Scholar] [CrossRef]
- Wanders, R.J.; Jansen, G.A.; Skjeldal, O.H. Refsum disease, peroxisomes and phytanic acid oxidation: A review. J. Neuropathol. Exp. Neurol. 2001, 60, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Branham, K.; Schlegel, D.; Pennesi, M.E.; Michaelides, M.; Heckenlively, J.; Jayasundera, T. PHYH. In Retinal Dystrophy Gene Atlas; Springer: Berlin/Heidelberg, Germany, 2018; p. 185. [Google Scholar]
- Waterham, H.R.; Wanders, R.J.A.; Leroy, B.P. Adult Refsum Disease. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mihalik, S.J.; Rainville, A.M.; Watkins, P.A. Phytanic acid alpha-oxidation in rat liver peroxisomes. Production of alpha-hydroxyphytanoyl-CoA and formate is enhanced by dioxygenase cofactors. Eur. J. Biochem. 1995, 232, 545–551. [Google Scholar] [CrossRef]
- Jansen, G.A.; Mihalik, S.J.; Watkins, P.A.; Moser, H.W.; Jakobs, C.; Denis, S.; Wanders, R.J. Phytanoyl-CoA hydroxylase is present in human liver, located in peroxisomes, and deficient in Zellweger syndrome: Direct, unequivocal evidence for the new, revised pathway of phytanic acid alpha-oxidation in humans. Biochem. Biophys. Res. Commun. 1996, 229, 205–210. [Google Scholar] [CrossRef]
- Molzer, B.; Stockler, S.; Bernheimer, H. Peroxisomal neurologic diseases and Refsum disease: Very long chain fatty acids and phytanic acid as diagnostic markers. Wien. Klin. Wochenschr. 1992, 104, 665–670. [Google Scholar] [PubMed]
- Skjeldal, O.H.; Stokke, O.; Refsum, S.; Norseth, J.; Petit, H. Clinical and biochemical heterogeneity in conditions with phytanic acid accumulation. J. Neurol. Sci. 1987, 77, 87–96. [Google Scholar] [CrossRef]
- Jansen, G.A.; Ofman, R.; Ferdinandusse, S.; Ijlst, L.; Muijsers, A.O.; Skjeldal, O.H.; Stokke, O.; Jakobs, C.; Besley, G.T.; Wraith, J.E.; et al. Refsum disease is caused by mutations in the phytanoyl-CoA hydroxylase gene. Nat. Genet. 1997, 17, 190–193. [Google Scholar] [CrossRef]
- Harari, D.; Gibberd, F.B.; Dick, J.P.; Sidey, M.C. Plasma exchange in the treatment of Refsum’s disease (heredopathia atactica polyneuritiformis). J. Neurol. Neurosurg. Psychiatry 1991, 54, 614–617. [Google Scholar] [CrossRef]
- Gibberd, F.B.; Billimoria, J.D.; Goldman, J.M.; Clemens, M.E.; Evans, R.; Whitelaw, M.N.; Retsas, S.; Sherratt, R.M. Heredopathia atactica polyneuritiformis: Refsum’s disease. Acta Neurol. Scand. 1985, 72, 1–17. [Google Scholar] [CrossRef]
- Srikajon, J.; Siritho, S.; Ngamsombat, C.; Prayoonwiwat, N.; Chirapapaisan, N.; Siriraj Neuroimmunology Research, G. Differences in clinical features between optic neuritis in neuromyelitis optica spectrum disorders and in multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 2055217318791196. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar] [CrossRef]
- Shosha, E.; Dubey, D.; Palace, J.; Nakashima, I.; Jacob, A.; Fujihara, K.; Takahashi, T.; Whittam, D.; Leite, M.I.; Misu, T.; et al. Area postrema syndrome: Frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology 2018, 91, e1642–e1651. [Google Scholar] [CrossRef] [PubMed]
- Brum, D.G.; Barreira, A.A.; dos Santos, A.C.; Kaimen-Maciel, D.R.; Matiello, M.; Costa, R.M.; Deghaide, N.H.; Costa, L.S.; Louzada-Junior, P.; Diniz, P.R.; et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult. Scler. 2010, 16, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Connell, C.M.; Janevic, M.R.; Gallant, M.P. The costs of caring: Impact of dementia on family caregivers. J. Geriatr. Psychiatry Neurol. 2001, 14, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. AQP4 antibodies in neuromyelitis optica: Diagnostic and pathogenetic relevance. Nat. Rev. Neurol. 2010, 6, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Nagelhus, E.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Patil, R.V.; Verkman, A.S. Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Invest. Ophthalmol. Vis. Sci. 2002, 43, 573–579. [Google Scholar]
- Jarius, S.; Franciotta, D.; Paul, F.; Ruprecht, K.; Bergamaschi, R.; Rommer, P.S.; Reuss, R.; Probst, C.; Kristoferitsch, W.; Wandinger, K.P.; et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J. Neuroinflammation 2010, 7, 52. [Google Scholar] [CrossRef]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef]
- Ratelade, J.; Verkman, A.S. Neuromyelitis optica: Aquaporin-4 based pathogenesis mechanisms and new therapies. Int. J. Biochem. Cell Biol. 2012, 44, 1519–1530. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, B.G. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Eculizumab: A Review in Neuromyelitis Optica Spectrum Disorder. Drugs 2020, 80, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Jade, J.D.; Bansi, S.; Singhal, B. Rituximab in Neuromyelitis Optica Spectrum Disorders: Our Experience. Ann. Indian Acad. Neurol. 2017, 20, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, A.I.; Pasco, P.M.D. Efficacy and tolerability of azathioprine for neuromyelitis optica spectrum disorder: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2019, 33, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.Y.; Kim, S.H.; Hyun, J.W.; Joung, A.R.; Park, M.S.; Kim, B.J.; Kim, H.J. Mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorder. JAMA Neurol. 2014, 71, 1372–1378. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Qiu, W.; Ma, H.; Zhang, X.; Zhu, Z.; Yang, C.-S.; Jia, D.; Zhang, T.-X.; Yuan, M. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): An open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020, 19, 391–401. [Google Scholar] [CrossRef]
- Papadopoulos, S.; Christodoulidou, M.; Gerasimidis, T. Perioperative Use of Antibiotics in Intra-Abdominal Surgical Infections. Surg. Infect. 2010, 11, 535–544. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, J.; Li, R.; Zhang, B.; Wang, Y.; Zhong, X.; Shu, Y.; Chang, Y.; Qiu, W. Disease Course and Outcomes in Patients With the Limited Form of Neuromyelitis Optica Spectrum Disorders and Negative AQP4-IgG Serology at Disease Onset: A Prospective Cohort Study. J. Clin. Neurol. 2022, 18, 453–462. [Google Scholar] [CrossRef]
- Higginbotham, H.; Eom, T.Y.; Mariani, L.E.; Bachleda, A.; Hirt, J.; Gukassyan, V.; Cusack, C.L.; Lai, C.; Caspary, T.; Anton, E.S. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev. Cell 2012, 23, 925–938. [Google Scholar] [CrossRef]
- Thomas, S.; Cantagrel, V.; Mariani, L.; Serre, V.; Lee, J.E.; Elkhartoufi, N.; de Lonlay, P.; Desguerre, I.; Munnich, A.; Boddaert, N.; et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur. J. Hum. Genet. 2015, 23, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, Y.; Lipschutz, J.H.; Lobo, G.P. Zebrafish as models to study ciliopathies of the eye and kidney. Clin. Nephrol. Res. 2017, 1, 6–9. [Google Scholar] [PubMed]
- Humbert, M.C.; Weihbrecht, K.; Searby, C.C.; Li, Y.; Pope, R.M.; Sheffield, V.C.; Seo, S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 19691–19696. [Google Scholar] [CrossRef]
- Roosing, S.; Rosti, R.O.; Rosti, B.; de Vrieze, E.; Silhavy, J.L.; van Wijk, E.; Wakeling, E.; Gleeson, J.G. Identification of a homozygous nonsense mutation in KIAA0556 in a consanguineous family displaying Joubert syndrome. Hum. Genet. 2016, 135, 919–921. [Google Scholar] [CrossRef]
- Sanders, A.A.; de Vrieze, E.; Alazami, A.M.; Alzahrani, F.; Malarkey, E.B.; Sorusch, N.; Tebbe, L.; Kuhns, S.; van Dam, T.J.; Alhashem, A.; et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015, 16, 293. [Google Scholar] [CrossRef]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell. Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Ott, T.; Kaufmann, L.; Granzow, M.; Hinderhofer, K.; Bartram, C.R.; Theiss, S.; Seitz, A.; Paramasivam, N.; Schulz, A.; Moog, U.; et al. The Frog Xenopus as a Model to Study Joubert Syndrome: The Case of a Human Patient With Compound Heterozygous Variants in PIBF1. Front. Physiol. 2019, 10, 134. [Google Scholar] [CrossRef]
- Hebbar, M.; Kanthi, A.; Shukla, A.; Bielas, S.; Girisha, K.M. A biallelic 36-bp insertion in PIBF1 is associated with Joubert syndrome. J. Hum. Genet. 2018, 63, 935–939. [Google Scholar] [CrossRef]
- Morbidoni, V.; Agolini, E.; Slep, K.C.; Pannone, L.; Zuccarello, D.; Cassina, M.; Grosso, E.; Gai, G.; Salviati, L.; Dallapiccola, B.; et al. Biallelic mutations in the TOGARAM1 gene cause a novel primary ciliopathy. J. Med. Genet. 2021, 58, 526–533. [Google Scholar] [CrossRef]
- Jacoby, M.; Cox, J.J.; Gayral, S.; Hampshire, D.J.; Ayub, M.; Blockmans, M.; Pernot, E.; Kisseleva, M.V.; Compere, P.; Schiffmann, S.N.; et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 2009, 41, 1027–1031. [Google Scholar] [CrossRef]
- Valente, E.M.; Marsh, S.E.; Castori, M.; Dixon-Salazar, T.; Bertini, E.; Al-Gazali, L.; Gleeson, J.G. Distinguishing the four genetic causes of Jouberts syndrome–related disorders. Ann. Neurol. 2005, 57, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Shaag, A.; Zenvirt, S.; Erlich, Y.; Hannon, G.J.; Shanske, A.L.; Gomori, J.M.; Ekstein, J.; Elpeleg, O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am. J. Hum. Genet. 2010, 86, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, S.; Yu, M.; Hu, H. TMEM216 Deletion Causes Mislocalization of Cone Opsin and Rhodopsin and Photoreceptor Degeneration in Zebrafish. Invest. Ophthalmol. Vis. Sci. 2020, 61, 24. [Google Scholar] [CrossRef] [PubMed]
- Dixon-Salazar, T.; Silhavy, J.L.; Marsh, S.E.; Louie, C.M.; Scott, L.C.; Gururaj, A.; Al-Gazali, L.; Al-Tawari, A.A.; Kayserili, H.; Sztriha, L.; et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004, 75, 979–987. [Google Scholar] [CrossRef]
- Utsch, B.; Sayer, J.A.; Attanasio, M.; Hennies, H.C.; Pohl, M.; Omran, H.; Hildebrandt, F. Confirmation of the JBTS3 locus and identification of a new ahi1 gene mutation in Joubert syndrome (JS) type 3 with renal involvement–evidence for other JS-causing genes in this region? Neuropediatrics 2005, 36, 31. [Google Scholar] [CrossRef]
- Brooks, B.P.; Zein, W.M.; Thompson, A.H.; Mokhtarzadeh, M.; Doherty, D.A.; Parisi, M.; Glass, I.A.; Malicdan, M.C.; Vilboux, T.; Vemulapalli, M.; et al. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology 2018, 125, 1937–1952. [Google Scholar] [CrossRef]
- Valente, E.M.; Brancati, F.; Silhavy, J.L.; Castori, M.; Marsh, S.E.; Barrano, G.; Bertini, E.; Boltshauser, E.; Zaki, M.S.; Abdel-Aleem, A.; et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 2006, 59, 527–534. [Google Scholar] [CrossRef]
- Brancati, F.; Barrano, G.; Silhavy, J.L.; Marsh, S.E.; Travaglini, L.; Bielas, S.L.; Amorini, M.; Zablocka, D.; Kayserili, H.; Al-Gazali, L.; et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am. J. Hum. Genet. 2007, 81, 104–113. [Google Scholar] [CrossRef]
- Vilboux, T.; Doherty, D.; Glass, I.; Parisi, M.; Phelps, I.; Cullinane, A.; Zein, W.; Brooks, B.; Heller, T.; Soldatos, A.; et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet. Med. 2017, 19, 875–882. [Google Scholar] [CrossRef]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef]
- Valente, E.M.; Brancati, F.; Dallapiccola, B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur. J. Med. Genet. 2008, 51, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, L.; Brancati, F.; Attie-Bitach, T.; Audollent, S.; Bertini, E.; Kaplan, J.; Perrault, I.; Iannicelli, M.; Mancuso, B.; Rigoli, L.; et al. Expanding CEP290 mutational spectrum in ciliopathies. Am. J. Med. Genet. A 2009, 149A, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Travaglini, L.; Zablocka, D.; Boltshauser, E.; Accorsi, P.; Montagna, G.; Silhavy, J.L.; Barrano, G.; Bertini, E.; Emma, F.; et al. RPGRIP1L mutations are mainly associated with the cerebello-renal phenotype of Joubert syndrome-related disorders. Clin. Genet. 2008, 74, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Arts, H.H.; Doherty, D.; van Beersum, S.E.; Parisi, M.A.; Letteboer, S.J.; Gorden, N.T.; Peters, T.A.; Märker, T.; Voesenek, K.; Kartono, A. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007, 39, 882–888. [Google Scholar] [CrossRef]
- Parisi, M.A. Clinical and molecular features of Joubert syndrome and related disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151C, 326–340. [Google Scholar] [CrossRef]
- Noor, A.; Windpassinger, C.; Patel, M.; Stachowiak, B.; Mikhailov, A.; Azam, M.; Irfan, M.; Siddiqui, Z.K.; Naeem, F.; Paterson, A.D.; et al. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am. J. Hum. Genet. 2008, 82, 1011–1018. [Google Scholar] [CrossRef]
- Doherty, D.; Parisi, M.A.; Finn, L.S.; Gunay-Aygun, M.; Al-Mateen, M.; Bates, D.; Clericuzio, C.; Demir, H.; Dorschner, M.; van Essen, A.J.; et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet. 2010, 47, 8–21. [Google Scholar] [CrossRef]
- Ben-Salem, S.; Al-Shamsi, A.M.; Gleeson, J.G.; Ali, B.R.; Al-Gazali, L. Mutation spectrum of Joubert syndrome and related disorders among Arabs. Hum. Genome Var. 2014, 1, 14020. [Google Scholar] [CrossRef]
- Janecke, A.R.; Muller, T.; Gassner, I.; Kreczy, A.; Schmid, E.; Kronenberg, F.; Utermann, B.; Utermann, G. Joubert-like syndrome unlinked to known candidate loci. J. Pediatr. 2004, 144, 264–269. [Google Scholar] [CrossRef]
- Slaats, G.G.; Isabella, C.R.; Kroes, H.Y.; Dempsey, J.C.; Gremmels, H.; Monroe, G.R.; Phelps, I.G.; Duran, K.J.; Adkins, J.; Kumar, S.A.; et al. MKS1 regulates ciliary INPP5E levels in Joubert syndrome. J. Med. Genet. 2016, 53, 62–72. [Google Scholar] [CrossRef]
- Utsch, B.; Sayer, J.A.; Attanasio, M.; Pereira, R.R.; Eccles, M.; Hennies, H.C.; Otto, E.A.; Hildebrandt, F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr. Nephrol. 2006, 21, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Mougou-Zerelli, S.; Thomas, S.; Szenker, E.; Audollent, S.; Elkhartoufi, N.; Babarit, C.; Romano, S.; Salomon, R.; Amiel, J.; Esculpavit, C.; et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum. Mutat. 2009, 30, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Brancati, F.; Iannicelli, M.; Travaglini, L.; Mazzotta, A.; Bertini, E.; Boltshauser, E.; D’Arrigo, S.; Emma, F.; Fazzi, E.; Gallizzi, R.; et al. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum. Mutat. 2009, 30, E432–E442. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.M.; Crinnion, L.A.; Berry, I.R.; Harrison, S.M.; Lascelles, C.; Antanaviciute, A.; Charlton, R.S.; Dobbie, A.; Carr, I.M.; Bonthron, D.T. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med. Genet. 2016, 17, 1. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dolle, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Putoux, A.; Thomas, S.; Coene, K.L.; Davis, E.E.; Alanay, Y.; Ogur, G.; Uz, E.; Buzas, D.; Gomes, C.; Patrier, S.; et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011, 43, 601–606. [Google Scholar] [CrossRef]
- Ali, B.R.; Silhavy, J.L.; Akawi, N.A.; Gleeson, J.G.; Al-Gazali, L. A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J. Rare Dis. 2012, 7, 27. [Google Scholar] [CrossRef]
- Lewis, T.R.; Kundinger, S.R.; Pavlovich, A.L.; Bostrom, J.R.; Link, B.A.; Besharse, J.C. Cos2/Kif7 and Osm-3/Kif17 regulate onset of outer segment development in zebrafish photoreceptors through distinct mechanisms. Dev. Biol. 2017, 425, 176–190. [Google Scholar] [CrossRef]
- Niceta, M.; Dentici, M.L.; Ciolfi, A.; Marini, R.; Barresi, S.; Lepri, F.R.; Novelli, A.; Bertini, E.; Cappa, M.; Digilio, M.C.; et al. Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: A case report and literature review. BMC Pediatr. 2020, 20, 120. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, W.; Ren, H.; Zhang, Y.; Niu, Y.; Wu, D.; Jiang, L. Non-classic splicing mutation in the CPLANE1 (C5orf42) gene cause Joubert syndrome in a fetus with severe craniocerebral dysplasia. Eur. J. Med. Genet. 2021, 64, 104212. [Google Scholar] [CrossRef]
- Asadollahi, R.; Strauss, J.E.; Zenker, M.; Beuing, O.; Edvardson, S.; Elpeleg, O.; Strom, T.M.; Joset, P.; Niedrist, D.; Otte, C.; et al. Clinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through Sonic Hedgehog signaling. Eur. J. Hum. Genet. 2018, 26, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Joubert, M.; Eisenring, J.J.; Robb, J.P.; Andermann, F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969, 19, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, J.; Meng, Q.; Wang, B. Three Tctn proteins are functionally conserved in the regulation of neural tube patterning and Gli3 processing but not ciliogenesis and Hedgehog signaling in the mouse. Dev. Biol. 2017, 430, 156–165. [Google Scholar] [CrossRef]
- Casoni, F.; Croci, L.; Bosone, C.; D’Ambrosio, R.; Badaloni, A.; Gaudesi, D.; Barili, V.; Sarna, J.R.; Tessarollo, L.; Cremona, O.; et al. Zfp423/ZNF423 regulates cell cycle progression, the mode of cell division and the DNA-damage response in Purkinje neuron progenitors. Development 2017, 144, 3686–3697. [Google Scholar] [CrossRef]
- Casoni, F.; Croci, L.; Vincenti, F.; Podini, P.; Riba, M.; Massimino, L.; Cremona, O.; Consalez, G.G. ZFP423 regulates early patterning and multiciliogenesis in the hindbrain choroid plexus. Development 2020, 147, dev190173. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Y.; Dong, H.; Gong, S.; Wei, B.; Luo, M.; Wang, H.; Wu, X.; Liu, W.; Xu, X.; et al. Loss of Tctn3 causes neuronal apoptosis and neural tube defects in mice. Cell Death Dis. 2018, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bangs, F.; Paton, I.R.; Prescott, A.; James, J.; Davey, M.G.; Whitley, P.; Genikhovich, G.; Technau, U.; Burt, D.W.; et al. The Talpid3 gene (KIAA0586) encodes a centrosomal protein that is essential for primary cilia formation. Development 2009, 136, 655–664. [Google Scholar] [CrossRef]
- Huppke, P.; Wegener, E.; Bohrer-Rabel, H.; Bolz, H.J.; Zoll, B.; Gartner, J.; Bergmann, C. Tectonic gene mutations in patients with Joubert syndrome. Eur. J. Hum. Genet. 2015, 23, 616–620. [Google Scholar] [CrossRef]
- Roosing, S.; Romani, M.; Isrie, M.; Rosti, R.O.; Micalizzi, A.; Musaev, D.; Mazza, T.; Al-Gazali, L.; Altunoglu, U.; Boltshauser, E.; et al. Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J. Med. Genet. 2016, 53, 608–615. [Google Scholar] [CrossRef]
- Brooks, E.R.; Islam, M.T.; Anderson, K.V.; Zallen, J.A. Sonic hedgehog signaling directs patterned cell remodeling during cranial neural tube closure. eLife 2020, 9, e60234. [Google Scholar] [CrossRef]
- Beck, B.B.; Phillips, J.B.; Bartram, M.P.; Wegner, J.; Thoenes, M.; Pannes, A.; Sampson, J.; Heller, R.; Gobel, H.; Koerber, F.; et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum. Mutat. 2014, 35, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Innes, A.M.; Boycott, K.M.; Puffenberger, E.G.; Redl, D.; MacDonald, I.M.; Chudley, A.E.; Beaulieu, C.; Perrier, R.; Gillan, T.; Wade, A.; et al. A founder mutation in BBS2 is responsible for Bardet-Biedl syndrome in the Hutterite population: Utility of SNP arrays in genetically heterogeneous disorders. Clin. Genet. 2010, 78, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Ghadami, M.; Tomita, H.A.; Najafi, M.T.; Damavandi, E.; Farahvash, M.S.; Yamada, K.; Majidzadeh-A, K.; Niikawa, N. Bardet-Biedl syndrome type 3 in an Iranian family: Clinical study and confirmation of disease localization. Am. J. Med. Genet. 2000, 94, 433–437. [Google Scholar] [CrossRef]
- Scheidecker, S.; Hull, S.; Perdomo, Y.; Studer, F.; Pelletier, V.; Muller, J.; Stoetzel, C.; Schaefer, E.; Defoort-Dhellemmes, S.; Drumare, I.; et al. Predominantly Cone-System Dysfunction as Rare Form of Retinal Degeneration in Patients With Molecularly Confirmed Bardet-Biedl Syndrome. Am. J. Ophthalmol. 2015, 160, 364–372.e1. [Google Scholar] [CrossRef] [PubMed]
- Young, T.L.; Penney, L.; Woods, M.O.; Parfrey, P.S.; Green, J.S.; Hefferton, D.; Davidson, W.S. A fifth locus for Bardet-Biedl syndrome maps to chromosome 2q31. Am. J. Hum. Genet. 1999, 64, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef]
- Billingsley, G.; Vincent, A.; Deveault, C.; Heon, E. Mutational analysis of SDCCAG8 in Bardet-Biedl syndrome patients with renal involvement and absent polydactyly. Ophthalmic Genet. 2012, 33, 150–154. [Google Scholar] [CrossRef]
- Otto, E.A.; Hurd, T.W.; Airik, R.; Chaki, M.; Zhou, W.; Stoetzel, C.; Patil, S.B.; Levy, S.; Ghosh, A.K.; Murga-Zamalloa, C.A.; et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 2010, 42, 840–850. [Google Scholar] [CrossRef]
- Novas, R.; Cardenas-Rodriguez, M.; Irigoin, F.; Badano, J.L. Bardet-Biedl syndrome: Is it only cilia dysfunction? FEBS Lett. 2015, 589, 3479–3491. [Google Scholar] [CrossRef]
- Schaefer, E.; Lauer, J.; Durand, M.; Pelletier, V.; Obringer, C.; Claussmann, A.; Braun, J.J.; Redin, C.; Mathis, C.; Muller, J.; et al. Mesoaxial polydactyly is a major feature in Bardet-Biedl syndrome patients with LZTFL1 (BBS17) mutations. Clin. Genet. 2014, 85, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Scheidecker, S.; Etard, C.; Pierce, N.W.; Geoffroy, V.; Schaefer, E.; Muller, J.; Chennen, K.; Flori, E.; Pelletier, V.; Poch, O.; et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J. Med. Genet. 2014, 51, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Li, Y.; Alhashem, A.; Anazi, S.; Alkuraya, H.; Hashem, M.; Awaji, A.A.; Sogaty, S.; Alkharashi, A.; Alzahrani, S.; et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum. Mol. Genet. 2014, 23, 3307–3315. [Google Scholar] [CrossRef]
- Schaefer, E.; Delvallee, C.; Mary, L.; Stoetzel, C.; Geoffroy, V.; Marks-Delesalle, C.; Holder-Espinasse, M.; Ghoumid, J.; Dollfus, H.; Muller, J. Identification and Characterization of Known Biallelic Mutations in the IFT27 (BBS19) Gene in a Novel Family With Bardet-Biedl Syndrome. Front. Genet. 2019, 10, 21. [Google Scholar] [CrossRef]
- Heon, E.; Kim, G.; Qin, S.; Garrison, J.E.; Tavares, E.; Vincent, A.; Nuangchamnong, N.; Scott, C.A.; Slusarski, D.C.; Sheffield, V.C. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 2016, 25, 2283–2294. [Google Scholar] [CrossRef]
- Khan, A.O.; Decker, E.; Bachmann, N.; Bolz, H.J.; Bergmann, C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet. 2016, 37, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Alsters, S.I.M.; Abawi, O.; Waisfisz, Q.; Boon, E.M.J.; van den Akker, E.L.T.; van Haelst, M.M. Second case of Bardet-Biedl syndrome caused by biallelic variants in IFT74. Eur. J. Hum. Genet. 2020, 28, 943–946. [Google Scholar] [CrossRef]
- Mardy, A.H.; Hodoglugil, U.; Yip, T.; Slavotinek, A.M. Third case of Bardet-Biedl syndrome caused by a biallelic variant predicted to affect splicing of IFT74. Clin. Genet. 2021, 100, 93–99. [Google Scholar] [CrossRef]
- Iannaccone, A.; Mykytyn, K.; Persico, A.M.; Searby, C.C.; Baldi, A.; Jablonski, M.M.; Sheffield, V.C. Clinical evidence of decreased olfaction in Bardet-Biedl syndrome caused by a deletion in the BBS4 gene. Am. J. Med. Genet. A 2005, 132A, 343–346. [Google Scholar] [CrossRef]
- Katsanis, N.; Eichers, E.R.; Ansley, S.J.; Lewis, R.A.; Kayserili, H.; Hoskins, B.E.; Scambler, P.J.; Beales, P.L.; Lupski, J.R. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am. J. Hum. Genet. 2002, 71, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Stone, E.M.; Mykytyn, K.; Heckenlively, J.R.; Green, J.S.; Heon, E.; Musarella, M.A.; Parfrey, P.S.; Sheffield, V.C.; Biesecker, L.G. Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 15–16. [Google Scholar] [CrossRef]
- Harville, H.M.; Held, S.; Diaz-Font, A.; Davis, E.E.; Diplas, B.H.; Lewis, R.A.; Borochowitz, Z.U.; Zhou, W.; Chaki, M.; MacDonald, J.; et al. Identification of 11 novel mutations in eight BBS genes by high-resolution homozygosity mapping. J. Med. Genet. 2010, 47, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Ansley, S.J.; Badano, J.L.; Blacque, O.E.; Hill, J.; Hoskins, B.E.; Leitch, C.C.; Kim, J.C.; Ross, A.J.; Eichers, E.R.; Teslovich, T.M.; et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003, 425, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Al-Anazi, S.; Al-Abdi, L.; Hashem, M.; Alkuraya, H.; Alamr, M.; Sirelkhatim, M.O.; Al-Hassnan, Z.; Alkuraya, B.; Mohamed, J.Y.; et al. In search of triallelism in Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2012, 20, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.J.; Zhang, H.X.; Huang, N.; Wu, K.C.; Huang, X.F.; Huang, F.; Tong, Y.; Pang, C.P.; Qu, J.; Jin, Z.B. Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS ONE 2014, 9, e90599. [Google Scholar] [CrossRef]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef]
- Suspitsin, E.N.; Imyanitov, E.N. Bardet-Biedl Syndrome. Mol Syndromol 2016, 7, 62–71. [Google Scholar] [CrossRef]
- Saida, K.; Inaba, Y.; Hirano, M.; Satake, W.; Toda, T.; Suzuki, Y.; Sudo, A.; Noda, S.; Hidaka, Y.; Hirabayashi, K.; et al. A case of Bardet-Biedl syndrome complicated with intracranial hypertension in a Japanese child. Brain Dev. 2014, 36, 721–724. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Zhang, Q.; Siemiatkowska, A.M.; Liu, Q.; Place, E.; Falk, M.J.; Consugar, M.; Lancelot, M.E.; Antonio, A.; Lonjou, C.; et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet. 2015, 24, 230–242. [Google Scholar] [CrossRef]
- Zhuang, J.C.; Huang, Z.Y.; Zhao, G.X.; Yu, H.; Li, Z.X.; Wu, Z.Y. Variants of CYP27B1 are associated with both multiple sclerosis and neuromyelitis optica patients in Han Chinese population. Gene 2015, 557, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Noto, D.; Sano, S.; Tomizawa, Y.; Yokoyama, K.; Hattori, N.; Miyake, S. Dysregulated B cell differentiation towards antibody-secreting cells in neuromyelitis optica spectrum disorder. J. Neuroinflammation 2022, 19, 6. [Google Scholar] [CrossRef]
- Brill, L.; Lavon, I.; Vaknin-Dembinsky, A. Reduced expression of the IL7Ra signaling pathway in Neuromyelitis optica. J. Neuroimmunol. 2018, 324, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, M.; Wang, N.; Xia, F.; Du, F.; Liu, Z.; Wang, J.; Jin, J.; Jin, B.; Zhao, G.; et al. Expression of CD226 is upregulated on Tr1 cells from neuromyelitis optica spectrum disorder patients. Brain Behav. 2022, 12, e2623. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.C.; Simmonds, M.J. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Wang, Y.G.; Fan, P.; Wang, J.Q.; Shu, Y.Q.; Li, R.; Zhong, X.N.; Long, L.; Zhao, Z.H.; Li, C.X.; et al. Expression of HLA-DP in patients with neuromyelitis optica spectrum disorders. Zhonghua Yi Xue Za Zhi 2019, 99, 3574–3580. [Google Scholar] [CrossRef] [PubMed]
- Pache, F.; Ringelstein, M.; Aktas, O.; Kleiter, I.; Jarius, S.; Siebert, N.; Bellmann-Strobl, J.; Paul, F.; Ruprecht, K. C3 and C4 complement levels in AQP4-IgG-positive NMOSD and in MOGAD. J. Neuroimmunol. 2021, 360, 577699. [Google Scholar] [CrossRef]


| Type | Disorder 1 | Phenotype OMIM 2 Number | Subtypes | Gene or Susceptibility Locus | Chromosomal Location | Gene OMIM 2 Number | Protein | Molecular Level | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Ciliopathy | JBTS2 | See in Table S1 | Transition zone (TZ) SHH signaling basal body (BB) | [88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131] | |||||
| BBS2 | See in Table S2 | BBSome protein chaperonin complex IFT | [132,133] | ||||||
| Alstrom syndrome | 203,800 | - | ALMS1 | 2p13.1 | 606,844 | ALMS1 | unclear | [134] | |
| Demyelination | MS 2 | 126,200 | MS1 | HLA-DQB1 HLA-DRB1 | 6p21.32 | 604,305 142,857 | HLA class II histocompatibility antigen, DQ beta 1 chain HLA class II histocompatibility antigen, DRB1 beta chain | chronic inflammation | [79,135,136,137] |
| 612,594 | MS2 | MS2 | 10p15.1 | 612,594 | - | ||||
| 612,595 | MS3 | MS3 | 5p13.2 | 612,595 | - | ||||
| 612,596 | MS4 | MS4 | 1p36 | 612,596 | - | ||||
| 614,810 | MS5 | TNFRSF1A | 12p13.31 | 191,190 | Tumor necrosis factor receptor superfamily member 1A | ||||
| AxD 2 | 203,450 | - | GFAP | 17q21.31 | 137,780 | Glial fibrillary acidic protein | GFAP aggregates | [138,139] | |
| PMD 2 | 312,080 | - | PLP1 | Xq22.2 | 300,401 | Myelin proteolipid protein | PLP1 accumulation | [140] | |
| Transcriptional Deregulation | MWS 2 | 235,730 | - | ZEB2 | 2q22.3 | 605,802 | Zinc finger E-box-binding homeobox 2 | transcription repressor targeting 5′-CACCT sequences interaction with Smads, TGFβ, and NuRD complex | [141] |
| PTHS 2 | 610,954 | - | TCF4 | 18q21.2 | 602,272 | Transcription factor 4 | transcription of neurogenesis | [142] | |
| RTT 2 | 312,750 | - | MeCP2 | Xq28 | 300,005 | Methyl-CpG-binding protein 2 | DNA and histone methylation reader transcription factor | [143] | |
| CS 2 | - | CS type I | ERCC6 ERCC8 | 10q11.23 5q12.1 | - | CSB CSA | DNA repair | [144,145] | |
| CS type II | |||||||||
| CS type III | |||||||||
| XP/CS | ERCC2 ERCC4 ERCC5 | 19q13.32 16p13.12 13q33.1 | - | General transcription and DNA repair factor IIH helicase subunit XPD DNA repair endonuclease XPF DNA excision repair protein XPG | |||||
| ATR-X 2 | 301,040 | - | ATRX | Xq21.1 | 301,040 | Transcriptional regulator ATRX | depositting histone variant | [146] | |
| Compromised Preoxisome | ZSD 2 | - | - | PEX1~13 | - | - | - | peroxisome formation peroxisomal protein transport | [147] |
| RD 2 | - | ARD | PHYH PEX7 | 10p13 6q23.3 | - | phytanoyl-CoA hydroxylase type 2 peroxisomal targeting signal receptor | oxidation of phytanic acid | [148] | |
| IRD | PEX1~13 | - | - | - | |||||
| Channelopathy | NMOSD 2 | - | - | See in Table S3 | cytotoxicity related to T cell, complement, NK | [79,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175] | |||
| Disease 1 | Target Gene | Mutation | Cas9 Ortholog and Delivery | Editing Mechanism | Model | Main Results | Reference |
|---|---|---|---|---|---|---|---|
| BBS1 | BBS1 | M390R | AAV2/5 vectors | Insert between two ITRs | M390R/M390R mice | 24% to 32% transduction in retina higher b-wave amplitudes in 50% mice | [263] |
| RTT | MeCP2 | - | AAV9 | - | Mecp2 null mice | Transduction efficiency: ~2–4% neurons observed improvements in survival | [351] |
| MeCP3 | - | AAV9 | - | Mecp2 null male mice | Partial amelioration in the null mouse model via provision of exogenously derived MeCP2 | [351] | |
| MeCP4 | - | scAAV9 | - | Mecp2 stop mice Mecp2B null mice | Reversing symptoms by ectopic expression of MeCP2 in virus infecting peripheral tissue and multiple cell types within the CNS | [355] | |
| MeCP5 | - | AAV9 | - | Mecp2 KO mice | Mecp2 transgene correcting breathing deficits and improving survival | [356] | |
| MeCP6 | - | AAV9 | - | Mecp2 null mice Mecp2tm1.1Bird mice Mecp2T158M mice | Direct cerebroventricular injection into neonatal mice resulting in high transduction efficiency, increased survival and body weight, and an amelioration of RTT-like phenotypes | [357] | |
| MeCP7 | - | AAV9 | - | Mecp2−/y mice | Modified vector extending lifespan without rescuing behavior | [358] | |
| MeCP8 | R270X | SpCas9, T2A | repairing induced DSBs by HR | MECP2R270X iPSC | developing CRISPR/Cas9-mediated system modifying MECP2 locus | [354] | |
| MeCP9 | - | AAV9 | - | Mecp2−/y mice | Insertion of miRARE improving safety without compromising efficacy | [359] | |
| CS | ERCC6 | c.643G>T (p.E215X) | pCAG-mCherry-gRNA vector | replace mutation with ssODN | CS-iPSCs | Alleviation of aging defects and recovered DNA repair ability | [360] |
| NMOSD | CART-BCMA | — | lentiviral vector | Add scFv | - | - | [361] |
| adRP | Rho | S334ter | SpCas9, plasmid electroporation | Allele-specific knockdown by indel | S334ter-3 rats | Nine-fold increase in photoreceptor nuclei 53% Improvement in the optokinetic response | [362] |
| Mertk | 1.9 kB deletion (intron 1–exon 2) | SpCas9, two AAV8 or 9 vectors | HITI-mediated insertion | RCS rats | Electroretinogram showing improved rod and cone responses compared with untreated and HDR-treated controls | [363] | |
| Nrl | — | SpCas9, two AAV8 vectors | Knockdown by indel (reprogram rods to cone-like cells) | Rho−/− and rd10 mice | 25% increase in cone photoreceptor preservation and electroretinogram B waves amplitude by ~60% | [364] | |
| Rho | P23H | SpCas9, two AAV8 vectors | Allele-specific knockdown by indel and wild-type supplementation | RhoP23H/P23H and RhoP23H/+ knock-in mice | Preserved electroretinogram B-waves and outer nuclear layer thickness in Cas9-treated mice compared with mice only given gene supplementation | [365] | |
| Rho | P23H | SpCas9-VQR, plasmid electroporation | Allele-specific knockdown by indel | RhoP23H/+ knock-in mice | Increase in wild-type mRNA by ~20% compared with untreated control Delayed outer nuclear layer degeneration | [366] | |
| Pde6b | Y347X | SpCas9/RecA, plasmid electroporation | Induce HDR using sgRNA-targeted RecA | rd1 mice | Increased survival of rod photoreceptors five-fold compared with nontreated controls | [367] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, K.-J.; Wu, H.-Y.; Yarmishyn, A.A.; Li, C.-Y.; Hsiao, Y.-J.; Chi, Y.-C.; Lo, T.-C.; Dai, H.-J.; Yang, Y.-C.; Liu, D.-H.; et al. Genetics behind Cerebral Disease with Ocular Comorbidity: Finding Parallels between the Brain and Eye Molecular Pathology. Int. J. Mol. Sci. 2022, 23, 9707. https://doi.org/10.3390/ijms23179707
Chang K-J, Wu H-Y, Yarmishyn AA, Li C-Y, Hsiao Y-J, Chi Y-C, Lo T-C, Dai H-J, Yang Y-C, Liu D-H, et al. Genetics behind Cerebral Disease with Ocular Comorbidity: Finding Parallels between the Brain and Eye Molecular Pathology. International Journal of Molecular Sciences. 2022; 23(17):9707. https://doi.org/10.3390/ijms23179707
Chicago/Turabian StyleChang, Kao-Jung, Hsin-Yu Wu, Aliaksandr A. Yarmishyn, Cheng-Yi Li, Yu-Jer Hsiao, Yi-Chun Chi, Tzu-Chen Lo, He-Jhen Dai, Yi-Chiang Yang, Ding-Hao Liu, and et al. 2022. "Genetics behind Cerebral Disease with Ocular Comorbidity: Finding Parallels between the Brain and Eye Molecular Pathology" International Journal of Molecular Sciences 23, no. 17: 9707. https://doi.org/10.3390/ijms23179707
APA StyleChang, K.-J., Wu, H.-Y., Yarmishyn, A. A., Li, C.-Y., Hsiao, Y.-J., Chi, Y.-C., Lo, T.-C., Dai, H.-J., Yang, Y.-C., Liu, D.-H., Hwang, D.-K., Chen, S.-J., Hsu, C.-C., & Kao, C.-L. (2022). Genetics behind Cerebral Disease with Ocular Comorbidity: Finding Parallels between the Brain and Eye Molecular Pathology. International Journal of Molecular Sciences, 23(17), 9707. https://doi.org/10.3390/ijms23179707