

Phospholipid Scramblase 4 (PLSCR4) Regulates Adipocyte Differentiation via PIP3-Mediated AKT Activation

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PLSCR4 Is Closely Related to PLSCR1 and Highly Expressed in Adipose Tissue
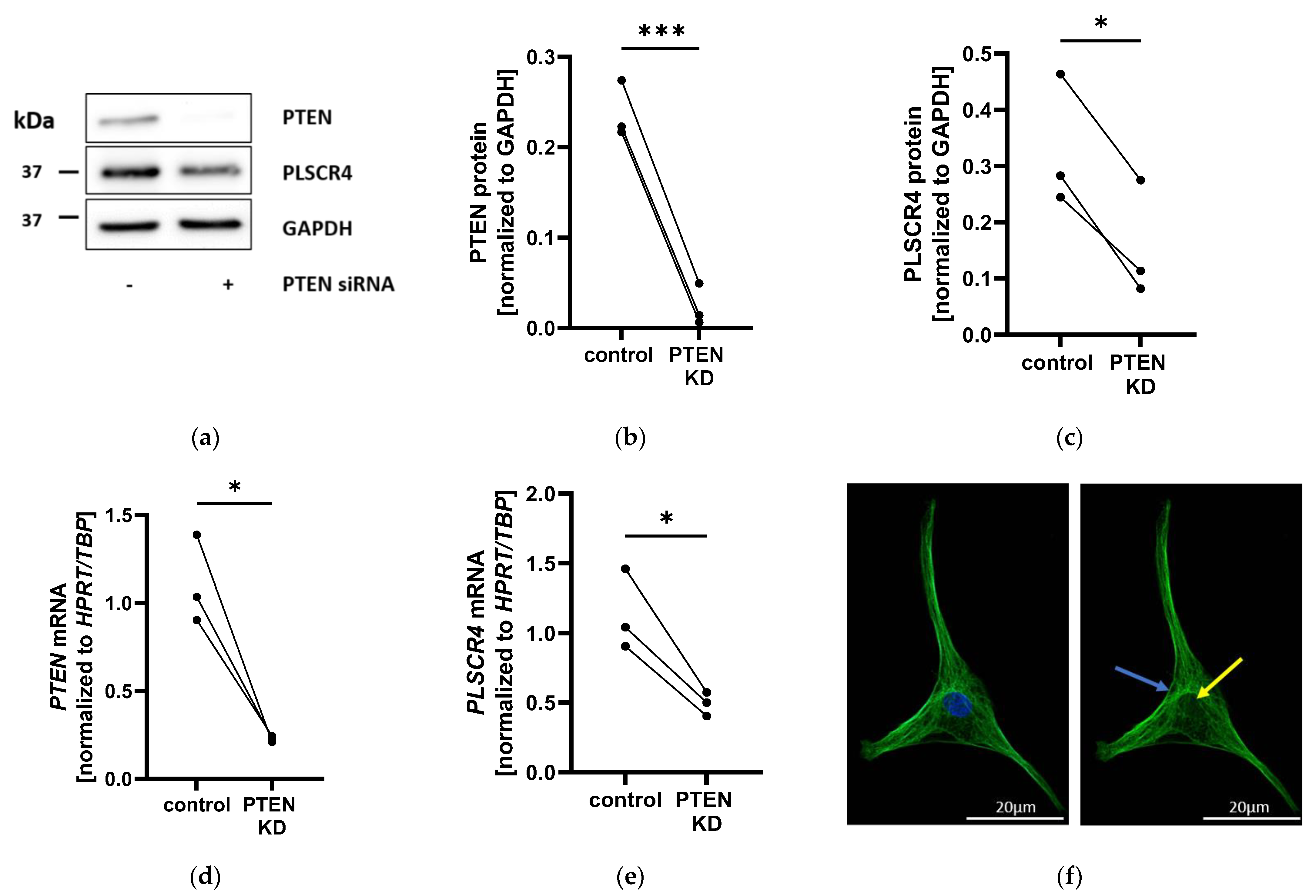
2.2. PLSCR4 Is Downregulated in PTEN-Knockdown Adipocyte Progenitor Cells and Localized on the Plasma Membrane
2.3. PLSCR4 Expression Is Altered in Different Models of Lipid Accumulation
2.4. Knockdown and Overexpression of PLSCR4 Affect Differentiation of Adipocyte Progenitor Cells
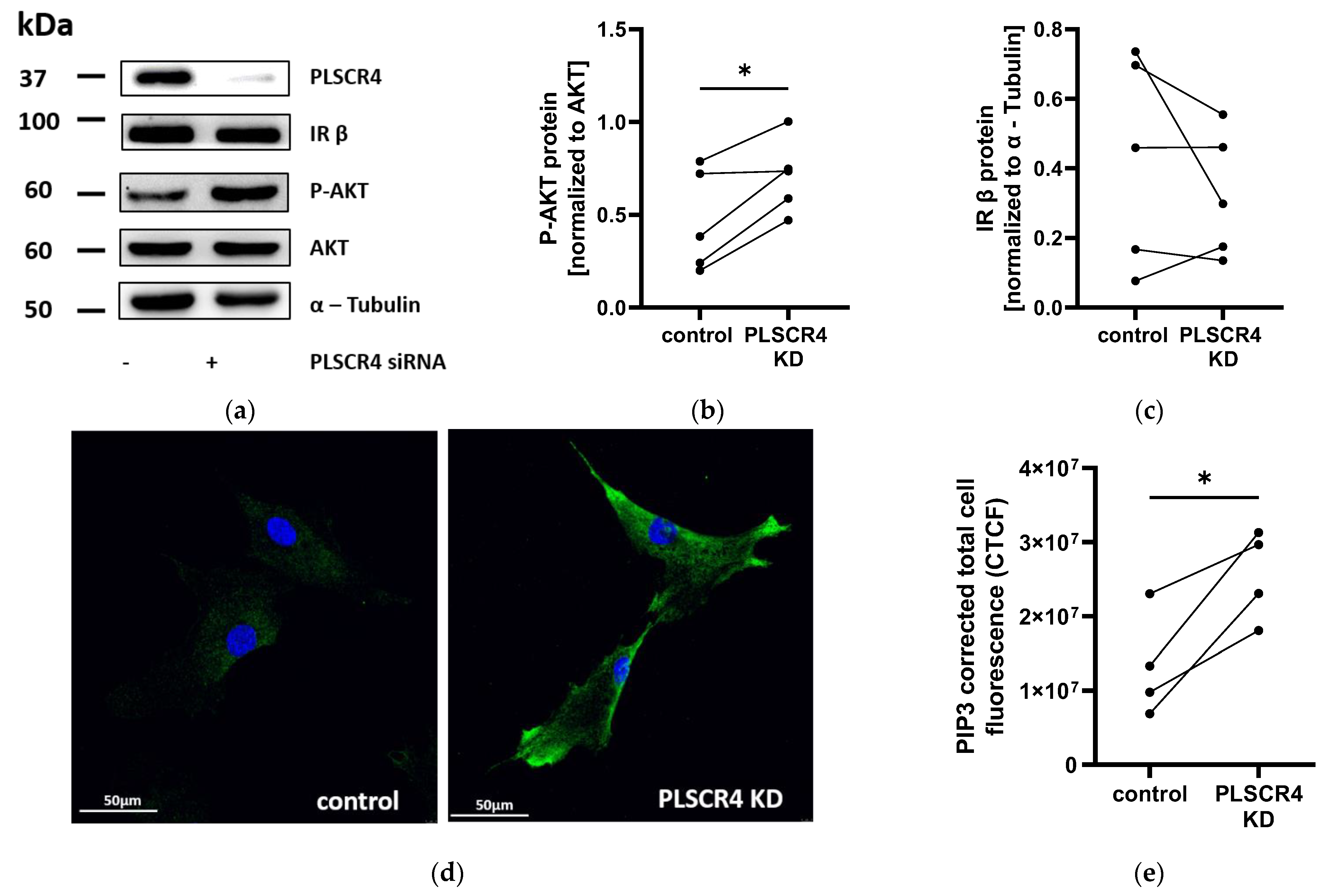
2.5. AKT Phosphorylation and PIP3 Immunofluorescence Staining Are Increased in PLSCR4 KD APCs
3. Discussion
4. Materials and Methods
4.1. PLSCR Gene Synteny and Expression Patterns
4.2. Cell Culture and In Vitro Adipocyte Differentiation
4.3. Quantitative Real-Time PCR (qPCR)
4.4. PLSCR4 Expression Analysis in Mouse Lipoma and White Adipose Tissue
4.5. PLSCR4 and PTEN siRNA Knockdown
4.6. PLSCR4 Overexpression Mediated by Plasmids
4.7. Proliferation Assays
4.8. Adipogenesis Assays
4.9. Immunofluorescence Staining
4.10. Western Blot Analysis
4.11. PIP2/PIP3 Quantification via High-Performance Liquid Chromatography–Mass Spectrometry (HPLC-MS)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Drissen, M.M.C.M.; Schieving, J.H.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R.; Hoogerbrugge, N. Red flags for early recognition of adult patients with PTEN Hamartoma Tumour Syndrome. Eur. J. Med. Genet. 2021, 64, 104364. [Google Scholar] [CrossRef]
- Schmid, G.L.; Kässner, F.; Uhlig, H.H.; Körner, A.; Kratzsch, J.; Händel, N.; Zepp, F.-P.; Kowalzik, F.; Laner, A.; Starke, S.; et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatr. Res. 2014, 75, 527–534. [Google Scholar] [CrossRef]
- Kirstein, A.S.; Kehr, S.; Nebe, M.; Hanschkow, M.; Barth, L.A.G.; Lorenz, J.; Penke, M.; Breitfeld, J.; Le Duc, D.; Landgraf, K.; et al. PTEN regulates adipose progenitor cell growth, differentiation, and replicative aging. J. Biol. Chem. 2021, 297, 100968. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, D.; Lin, C.-C.; Popkova, Y.; Yang, Z.; Akhil, V.; Çakir, M.V.; Grunewald, S.; Simon, J.-C.; Dietz, A.; Dannenberger, D.; et al. Reduced lipolysis in lipoma phenocopies lipid accumulation in obesity. Int. J. Obes. 2021, 45, 565–576. [Google Scholar] [CrossRef]
- Breitfeld, J.; Kehr, S.; Müller, L.; Stadler, P.F.; Böttcher, Y.; Blüher, M.; Stumvoll, M.; Kovacs, P. Developmentally Driven Changes in Adipogenesis in Different Fat Depots Are Related to Obesity. Front. Endocrinol. 2020, 11, 138. [Google Scholar] [CrossRef]
- Wiedmer, T.; Zhou, Q.; Kwoh, D.Y.; Sims, P.J. Identification of three new members of the phospholipid scramblase gene family. Biochim. Biophys. Acta Biomembr. 2000, 1467, 244–253. [Google Scholar] [CrossRef]
- Falzone, M.E.; Accardi, A. Reconstitution of Proteoliposomes for Phospholipid Scrambling and Nonselective Channel Assays. Methods Mol. Biol. 2020, 2127, 207–225. [Google Scholar] [CrossRef]
- Kodigepalli, K.M.; Bowers, K.; Sharp, A.; Nanjundan, M. Roles and regulation of phospholipid scramblases. FEBS Lett. 2015, 589, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Wiedmer, T.; Zhao, J.; Li, L.; Zhou, Q.; Hevener, A.; Olefsky, J.M.; Curtiss, L.K.; Sims, P.J. Adiposity, dyslipidemia, and insulin resistance in mice with targeted deletion of phospholipid scramblase 3 (PLSCR3). Proc. Natl. Acad. Sci. USA 2004, 101, 13296–13301. [Google Scholar] [CrossRef] [PubMed]
- Kerner, B.; Rao, A.R.; Christensen, B.; Dandekar, S.; Yourshaw, M.; Nelson, S.F. Rare Genomic Variants Link Bipolar Disorder with Anxiety Disorders to CREB-Regulated Intracellular Signaling Pathways. Front. Psychiatry 2013, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, D.; Zhang, X.; Lv, M.; Liu, G.; Gu, C.; Yang, F.; Wang, Y. Effect and mechanism of phospholipid scramblase 4 (PLSCR4) on lipopolysaccharide (LPS)-induced injury to human pulmonary microvascular endothelial cells. Ann. Transl. Med. 2021, 9, 159. [Google Scholar] [CrossRef]
- Abdulwahab, H.; Aljishi, M.; Sultan, A.; Al-Kafaji, G.; Sridharan, K.; Bakhiet, M.; Taha, S. Whole blood transcriptomic analysis reveals PLSCR4 as a potential marker for vaso-occlusive crises in sickle cell disease. Sci. Rep. 2021, 11, 22199. [Google Scholar] [CrossRef]
- Song, Y.-B.; An, Y.R.; Kim, S.J.; Park, H.-W.; Jung, J.-W.; Kyung, J.-S.; Hwang, S.Y.; Kim, Y.-S. Lipid metabolic effect of Korean red ginseng extract in mice fed on a high-fat diet. J. Sci. Food Agric. 2012, 92, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Graves, J.A.M.; Rens, W. The evolution of marsupial and monotreme chromosomes. Cytogenet. Genome Res. 2012, 137, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Laurenczy, B.; Gharib, W.H.; Castella, B.; Kuzniar, A.; Schabauer, H.; Studer, R.A.; Valle, M.; Salamin, N.; Stockinger, H.; et al. Selectome update: Quality control and computational improvements to a database of positive selection. Nucleic Acids Res. 2014, 42, D917–D921. [Google Scholar] [CrossRef]
- Velluva, A.; Radtke, M.; Horn, S.; Popp, B.; Platzer, K.; Gjermeni, E.; Lin, C.-C.; Lemke, J.R.; Garten, A.; Schöneberg, T.; et al. Phenotype-tissue expression and exploration (PTEE) resource facilitates the choice of tissue for RNA-seq-based clinical genetics studies. BMC Genom. 2021, 22, 802. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Filtz, E.A.; Emery, A.; Lu, H.; Forster, C.L.; Karasch, C.; Hallstrom, T.C. Rb1 and Pten Co-Deletion in Osteoblast Precursor Cells Causes Rapid Lipoma Formation in Mice. PLoS ONE 2015, 10, e0136729. [Google Scholar] [CrossRef] [Green Version]
- Kässner, F.; Kirstein, A.; Händel, N.; Schmid, G.L.; Landgraf, K.; Berthold, A.; Tannert, A.; Schaefer, M.; Wabitsch, M.; Kiess, W.; et al. A new human adipocyte model with PTEN haploinsufficiency. Adipocyte 2020, 9, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Ben-Efraim, I.; Zhou, Q.; Wiedmer, T.; Gerace, L.; Sims, P.J. Phospholipid scramblase 1 is imported into the nucleus by a receptor-mediated pathway and interacts with DNA. Biochemistry 2004, 43, 3518–3526. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nanjundan, M.; Pike, L.J.; Wiedmer, T.; Sims, P.J. Plasma membrane phospholipid scramblase 1 is enriched in lipid rafts and interacts with the epidermal growth factor receptor. Biochemistry 2002, 41, 6338–6345. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Guy, L.; Kultima, J.R.; Andersson, S.G.E. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2010, 26, 2334–2335. [Google Scholar] [CrossRef]
- Sanjabi, S.; Williams, K.J.; Saccani, S.; Zhou, L.; Hoffmann, A.; Ghosh, G.; Gerondakis, S.; Natoli, G.; Smale, S.T. A c-Rel subdomain responsible for enhanced DNA-binding affinity and selective gene activation. Genes Dev. 2005, 19, 2138–2151. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Fitzpatrick, M. Measuring Cell Fluorescence Using ImageJ—The Open Lab Book v1.0. Available online: https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html (accessed on 13 June 2022).
- Clark, J.; Anderson, K.E.; Juvin, V.; Smith, T.S.; Karpe, F.; Wakelam, M.J.O.; Stephens, L.R.; Hawkins, P.T. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 2011, 8, 267–272. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barth, L.A.G.; Nebe, M.; Kalwa, H.; Velluva, A.; Kehr, S.; Kolbig, F.; Prabutzki, P.; Kiess, W.; Le Duc, D.; Garten, A.; et al. Phospholipid Scramblase 4 (PLSCR4) Regulates Adipocyte Differentiation via PIP3-Mediated AKT Activation. Int. J. Mol. Sci. 2022, 23, 9787. https://doi.org/10.3390/ijms23179787
Barth LAG, Nebe M, Kalwa H, Velluva A, Kehr S, Kolbig F, Prabutzki P, Kiess W, Le Duc D, Garten A, et al. Phospholipid Scramblase 4 (PLSCR4) Regulates Adipocyte Differentiation via PIP3-Mediated AKT Activation. International Journal of Molecular Sciences. 2022; 23(17):9787. https://doi.org/10.3390/ijms23179787
Chicago/Turabian StyleBarth, Lisa A. G., Michèle Nebe, Hermann Kalwa, Akhil Velluva, Stephanie Kehr, Florentien Kolbig, Patricia Prabutzki, Wieland Kiess, Diana Le Duc, Antje Garten, and et al. 2022. "Phospholipid Scramblase 4 (PLSCR4) Regulates Adipocyte Differentiation via PIP3-Mediated AKT Activation" International Journal of Molecular Sciences 23, no. 17: 9787. https://doi.org/10.3390/ijms23179787
APA StyleBarth, L. A. G., Nebe, M., Kalwa, H., Velluva, A., Kehr, S., Kolbig, F., Prabutzki, P., Kiess, W., Le Duc, D., Garten, A., & Kirstein, A. S. (2022). Phospholipid Scramblase 4 (PLSCR4) Regulates Adipocyte Differentiation via PIP3-Mediated AKT Activation. International Journal of Molecular Sciences, 23(17), 9787. https://doi.org/10.3390/ijms23179787