
Breaking the Concentration Limit in Fluorescence Fluctuation Spectroscopy with Camera-Based Detection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
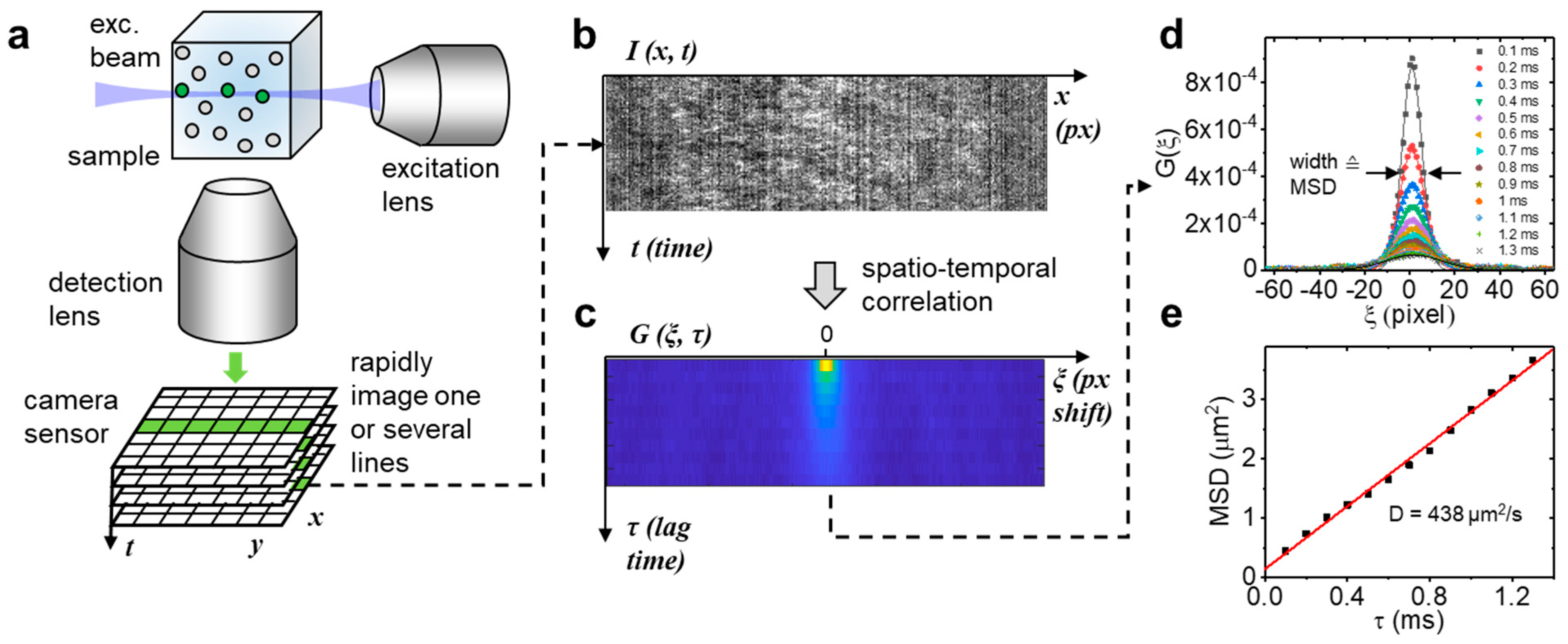
2.1. LIM Method Validation
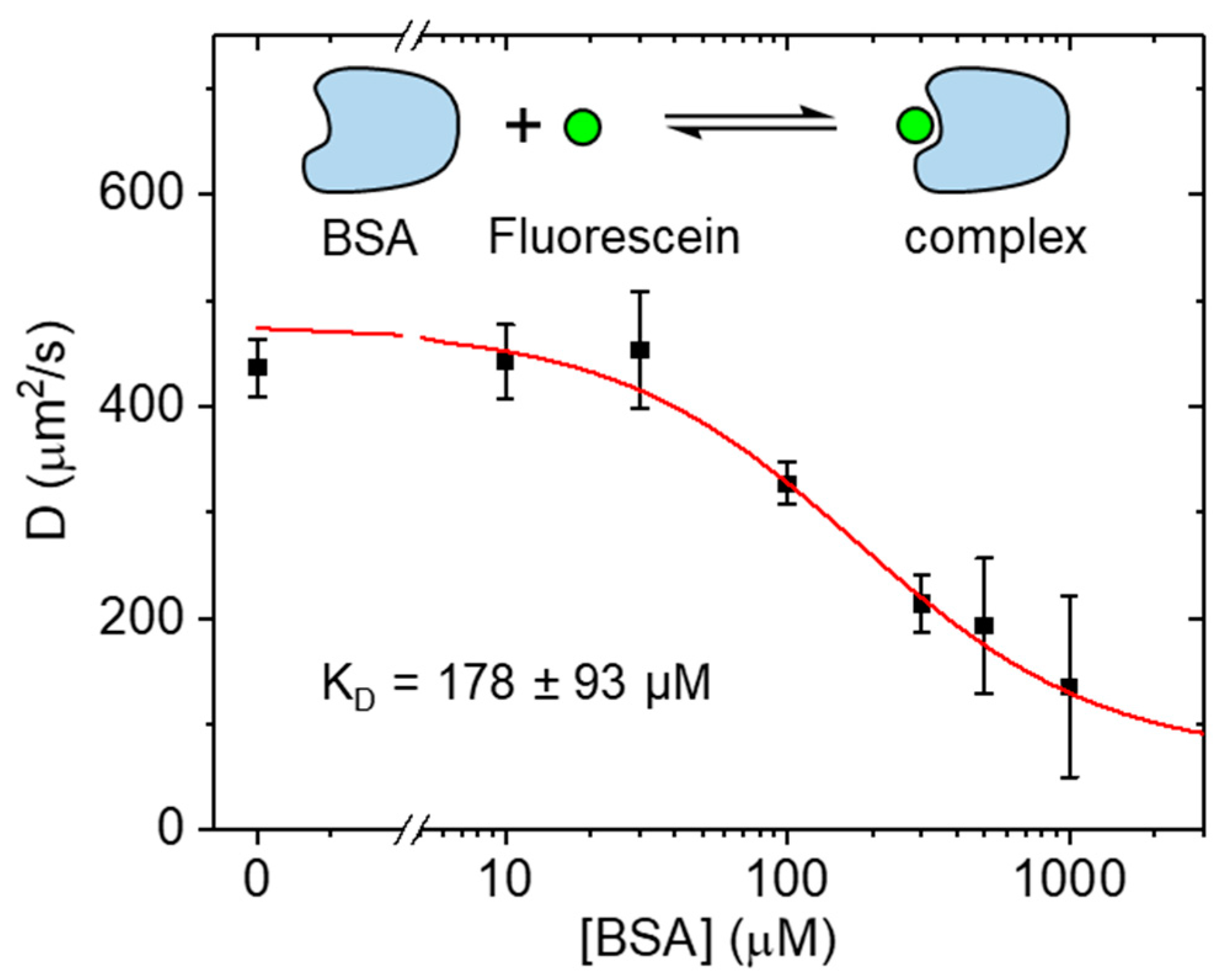
2.2. Binding at High Concentration
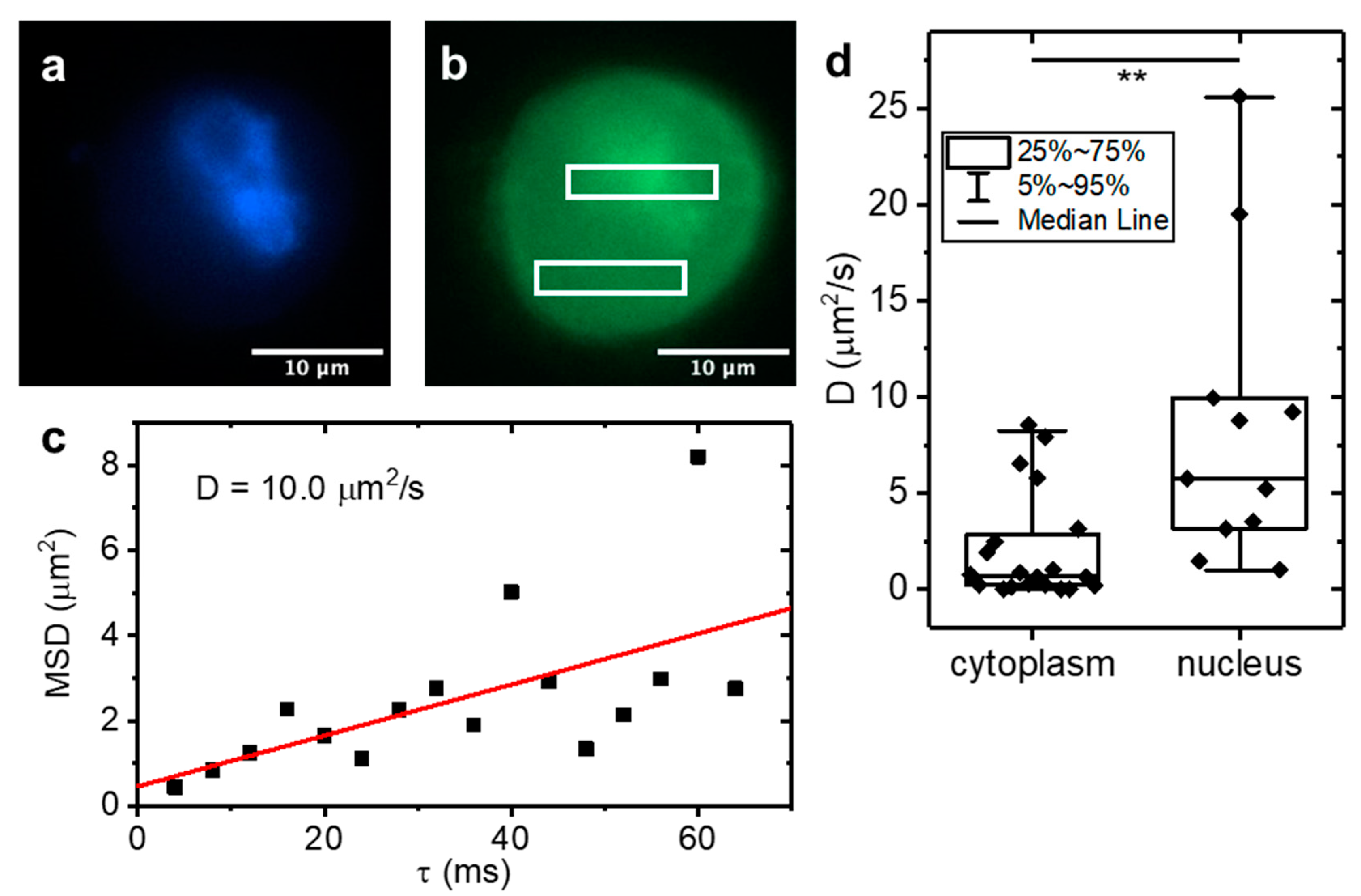
2.3. Application to Cells
3. Discussion
4. Methods and Materials
4.1. Line Illumination
4.2. Single Point FCS
4.3. Dye and Protein Solutions
4.4. Cell Sample Preparation
4.5. Measurement Parameters
4.6. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar] [CrossRef] [PubMed]
- Jameson, D.M.; Ross, J.A.; Albanesi, J.P. Fluorescence fluctuation spectroscopy: Ushering in a new age of enlightenment for cellular dynamics. Biophys. Rev. 2009, 1, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Su, D.; Shao, C.; Wang, J.; Dong, C.; Huang, X.; Ren, J. A sensitive and microscale method for drug screening combining affinity probes and single molecule fluorescence correlation spectroscopy. Analyst 2015, 140, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; Von Middendorff, C.; Schönle, A.; et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Wenger, J.; Lenne, P.-F.; Popov, E.; Rigneault, H.; Dintinger, J.; Ebbesen, T.W. Single molecule fluorescence in rectangular nano-apertures. Opt. Express 2005, 13, 7035–7044. [Google Scholar] [CrossRef] [PubMed]
- Leutenegger, M.; Gösch, M.; Perentes, A.; Hoffmann, P.; Martin, O.J.F.; Lasser, T. Confining the sampling volume for Fluorescence Correlation Spectroscopy using a sub-wavelength sized aperture. Opt. Express 2006, 14, 956–969. [Google Scholar] [CrossRef]
- Vobornik, D.; Banks, D.S.; Lu, Z.; Fradin, Ć.; Taylor, R.; Johnston, L.J. Fluorescence correlation spectroscopy with sub-diffraction-limited resolution using near-field optical probes. Appl. Phys. Lett. 2008, 93, 163904. [Google Scholar] [CrossRef]
- Koppel, D.E. Statistical accuracy in fluorescence correlation spectroscopy. Phys. Rev. A 1974, 10, 1938–1945. [Google Scholar] [CrossRef]
- Laurence, T.A.; Ly, S.; Bourguet, F.; Fischer, N.O.; Coleman, M.A. Fluorescence correlation spectroscopy at micromolar concentrations without optical nanoconfinement. J. Phys. Chem. B 2014, 118, 9662–9667. [Google Scholar] [CrossRef] [PubMed]
- Hedde, P.N.; Malacrida, L.; Ahrar, S.; Siryaporn, A.; Gratton, E. sideSPIM—Selective plane illumination based on a conventional inverted microscope. Biomed. Opt. Express 2017, 8, 3918–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedde, P.N.; Stakic, M.; Gratton, E. Rapid measurement of molecular transport and interaction inside living cells using single plane illumination. Sci. Rep. 2014, 4, 7048. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, C.; Gratton, E.; Beltram, F.; Cardarelli, F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. USA 2013, 110, 12307–12312. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, P. Absolute Diffusion Coefficients: Compilation of Reference Data for FCS Calibration. Available online: https://www.picoquant.com/images/uploads/page/files/7353/appnote_diffusioncoefficients.pdf (accessed on 10 July 2020).
- Mao, Y.; Yu, L.; Yang, R.; Qu, L.B.; Harrington, P.D.B. A novel method for the study of molecular interaction by using microscale thermophoresis. Talanta 2015, 132, 894–901. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Levi, V.; González Flecha, F.L. Reversible fast-dimerization of bovine serum albumin detected by fluorescence resonance energy transfer. Biochim. Biophys. Acta—Proteins Proteom. 2002, 1599, 141–148. [Google Scholar] [CrossRef]
- Hedde, P.N.; Dörlich, R.M.; Blomley, R.; Gradl, D.; Oppong, E.; Cato, A.C.B.; Nienhaus, G.U. Stimulated emission depletion-based raster image correlation spectroscopy reveals biomolecular dynamics in live cells. Nat. Commun. 2013, 4, 2093. [Google Scholar] [CrossRef] [PubMed]
- Hedde, P.N.; Barylko, B.; Binns, D.D.; Jameson, D.M.; Albanesi, J.P. Differential Mobility and Self-Association of Arc/Arg3.1 in the Cytoplasm and Nucleus of Living Cells. ACS Chem. Neurosci. 2022, 13, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Gendron, P.O.; Avaltroni, F.; Wilkinson, K.J. Diffusion coefficients of several rhodamine derivatives as determined by pulsed field gradient-nuclear magnetic resonance and fluorescence correlation spectroscopy. J. Fluoresc. 2008, 18, 1093–1101. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-K.; Hedde, P.N. Breaking the Concentration Limit in Fluorescence Fluctuation Spectroscopy with Camera-Based Detection. Int. J. Mol. Sci. 2022, 23, 9840. https://doi.org/10.3390/ijms23179840
Huang Y-K, Hedde PN. Breaking the Concentration Limit in Fluorescence Fluctuation Spectroscopy with Camera-Based Detection. International Journal of Molecular Sciences. 2022; 23(17):9840. https://doi.org/10.3390/ijms23179840
Chicago/Turabian StyleHuang, Yu-Kai, and Per Niklas Hedde. 2022. "Breaking the Concentration Limit in Fluorescence Fluctuation Spectroscopy with Camera-Based Detection" International Journal of Molecular Sciences 23, no. 17: 9840. https://doi.org/10.3390/ijms23179840
APA StyleHuang, Y.-K., & Hedde, P. N. (2022). Breaking the Concentration Limit in Fluorescence Fluctuation Spectroscopy with Camera-Based Detection. International Journal of Molecular Sciences, 23(17), 9840. https://doi.org/10.3390/ijms23179840