
Atropine Is a Suppressor of Epithelial–Mesenchymal Transition (EMT) That Reduces Stemness in Drug-Resistant Breast Cancer Cells


Abstract
:1. Introduction
2. Results
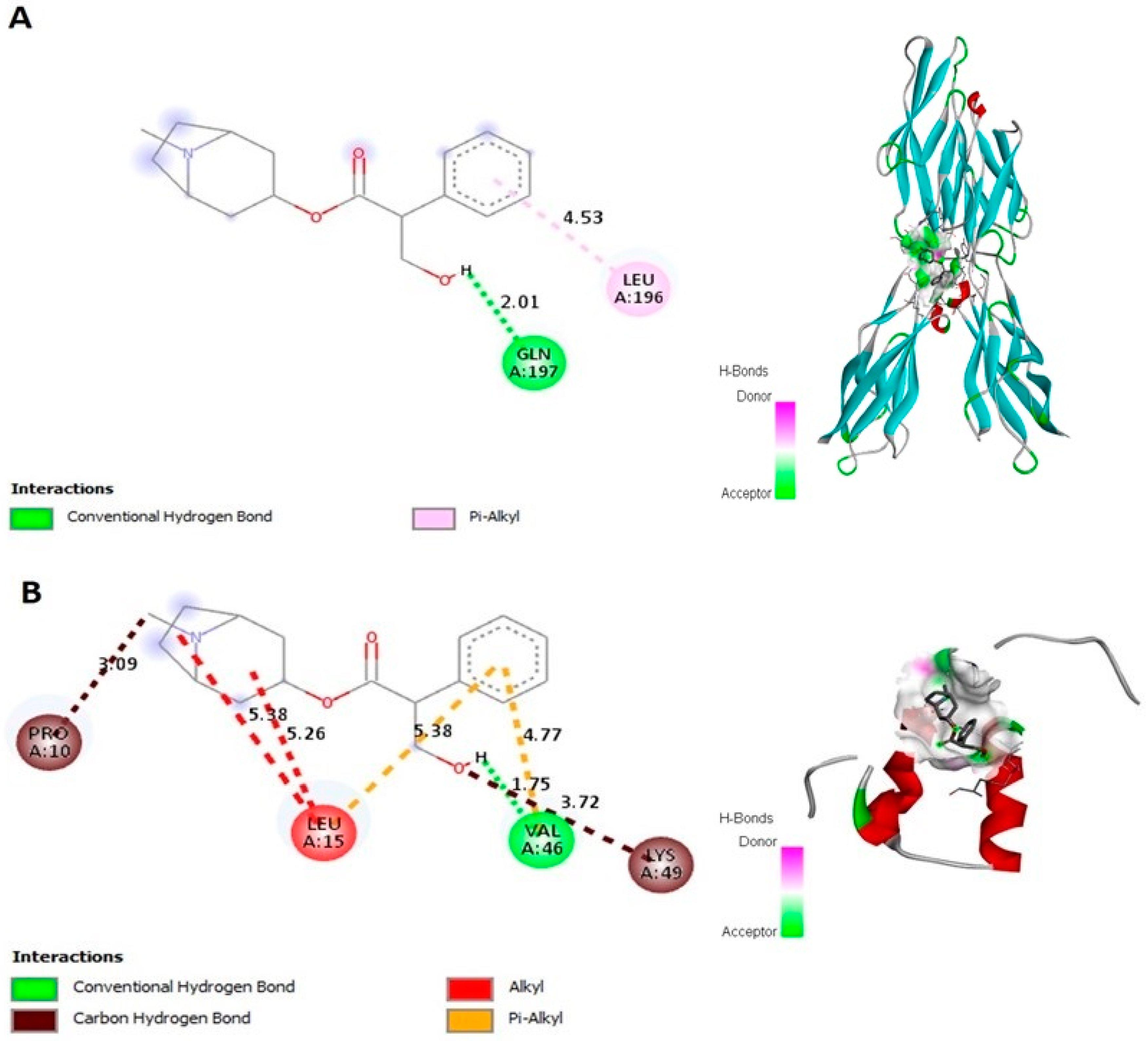
2.1. Atropine Interacts with E-cad and Zeb-2 In Silico
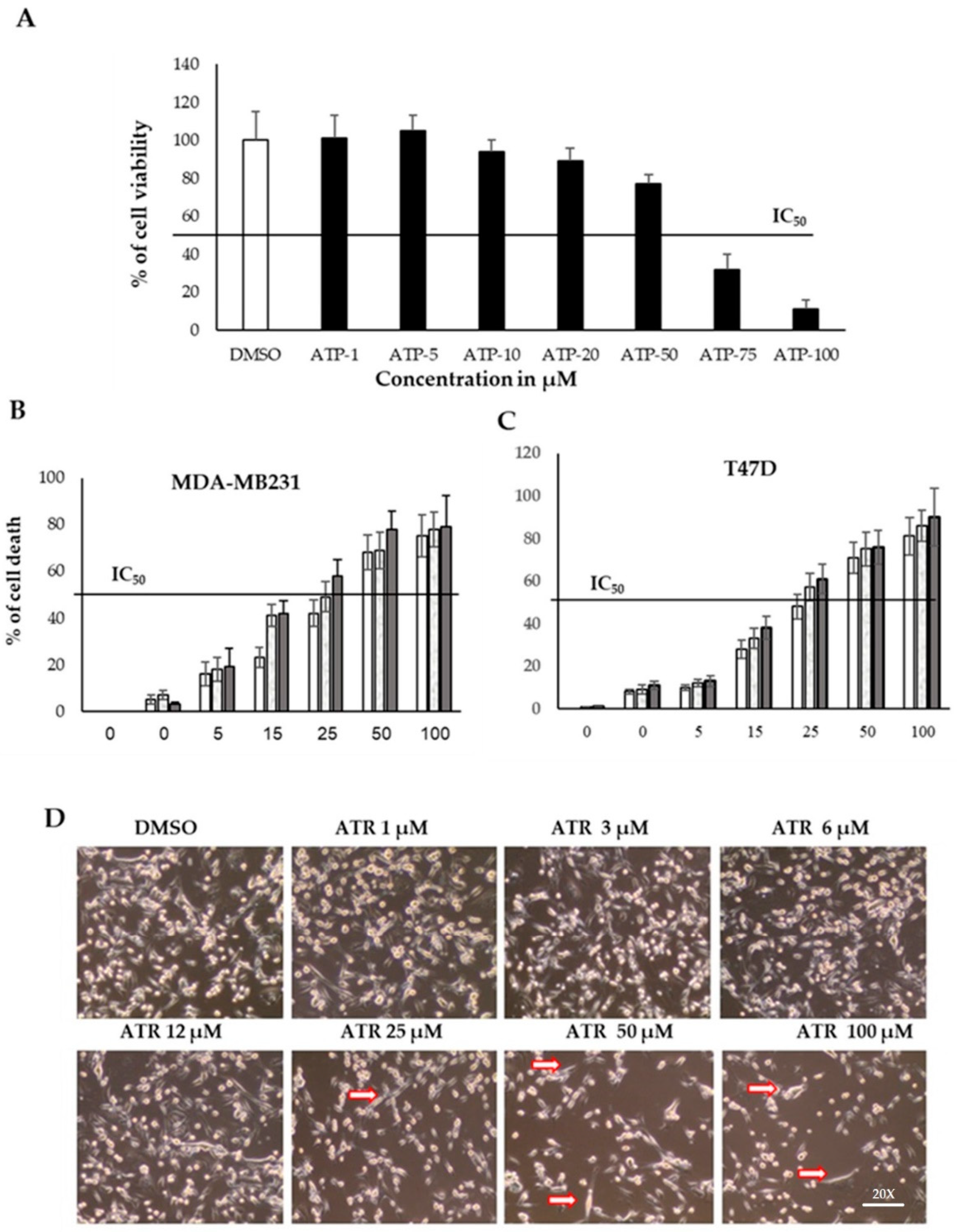
2.2. Atropine Reduces the Proliferation Rate in Breast Cancer Cell Lines
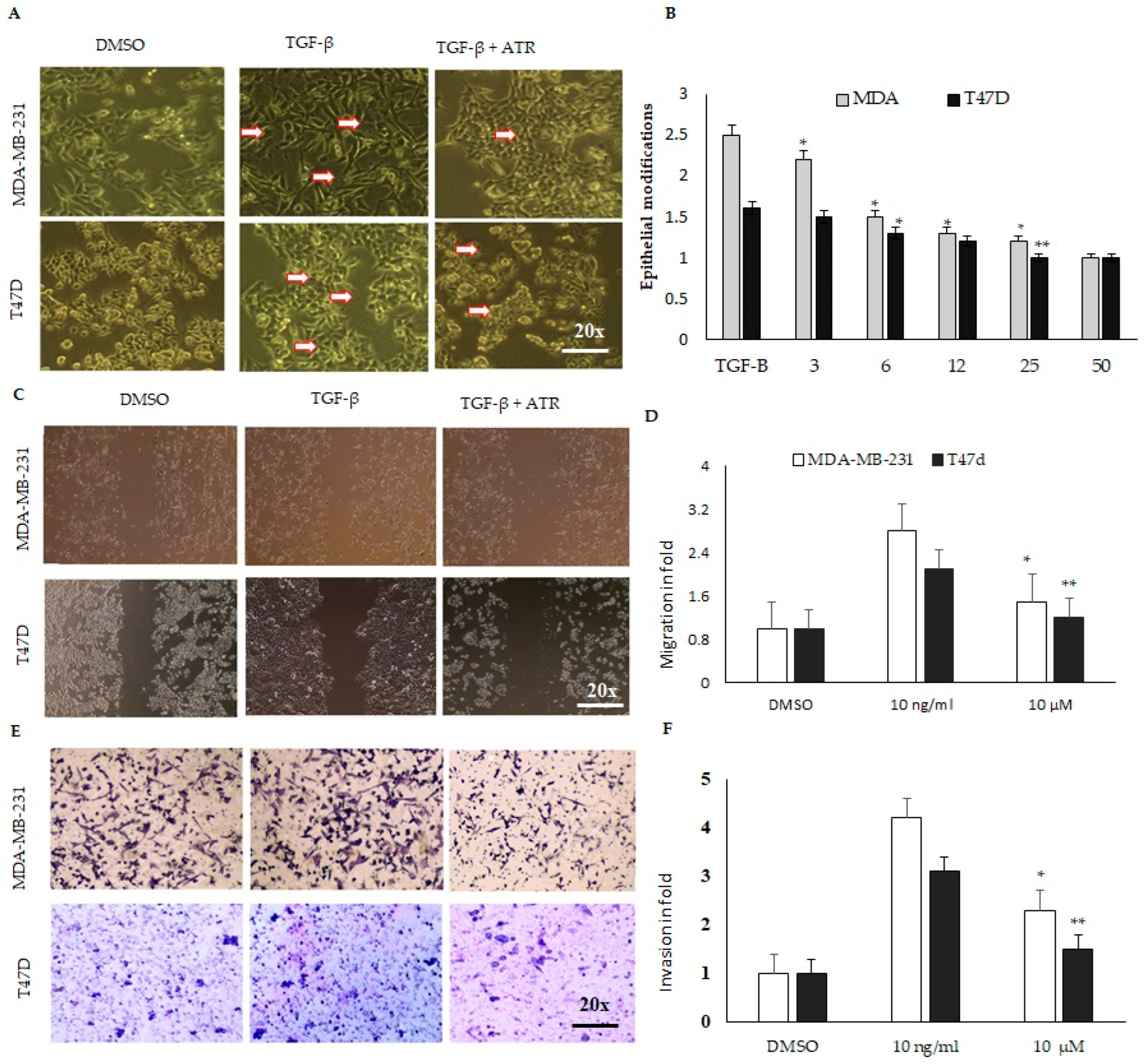
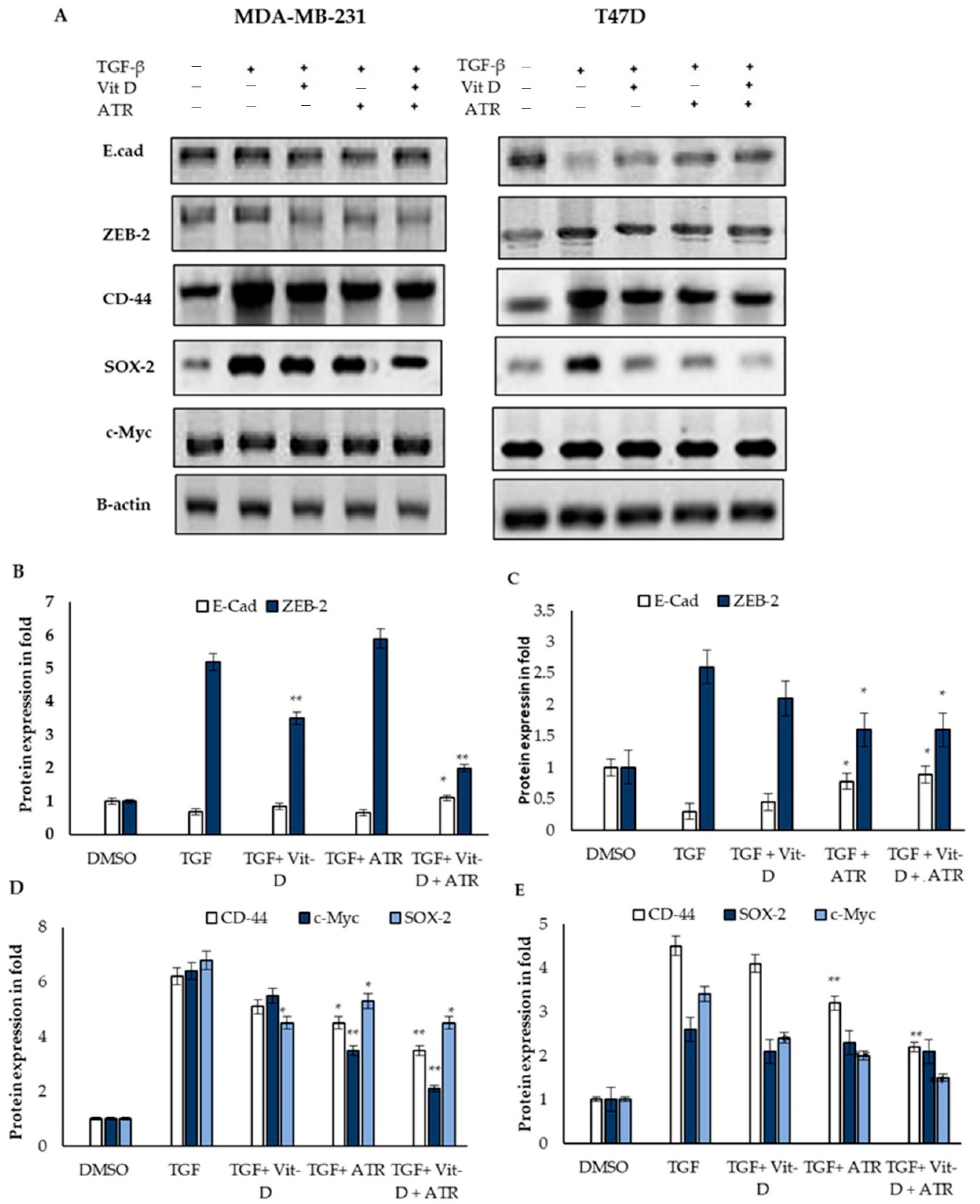
2.3. Atropine Suppresses EMT in MDA-MB-231 and T47D Cancer Cell Lines
2.4. Atropine Abrogates the Cancer Stem Cell Modifications and Suppresses Migration and Invasion in MDA-MB-231 and T47D Cell Lines
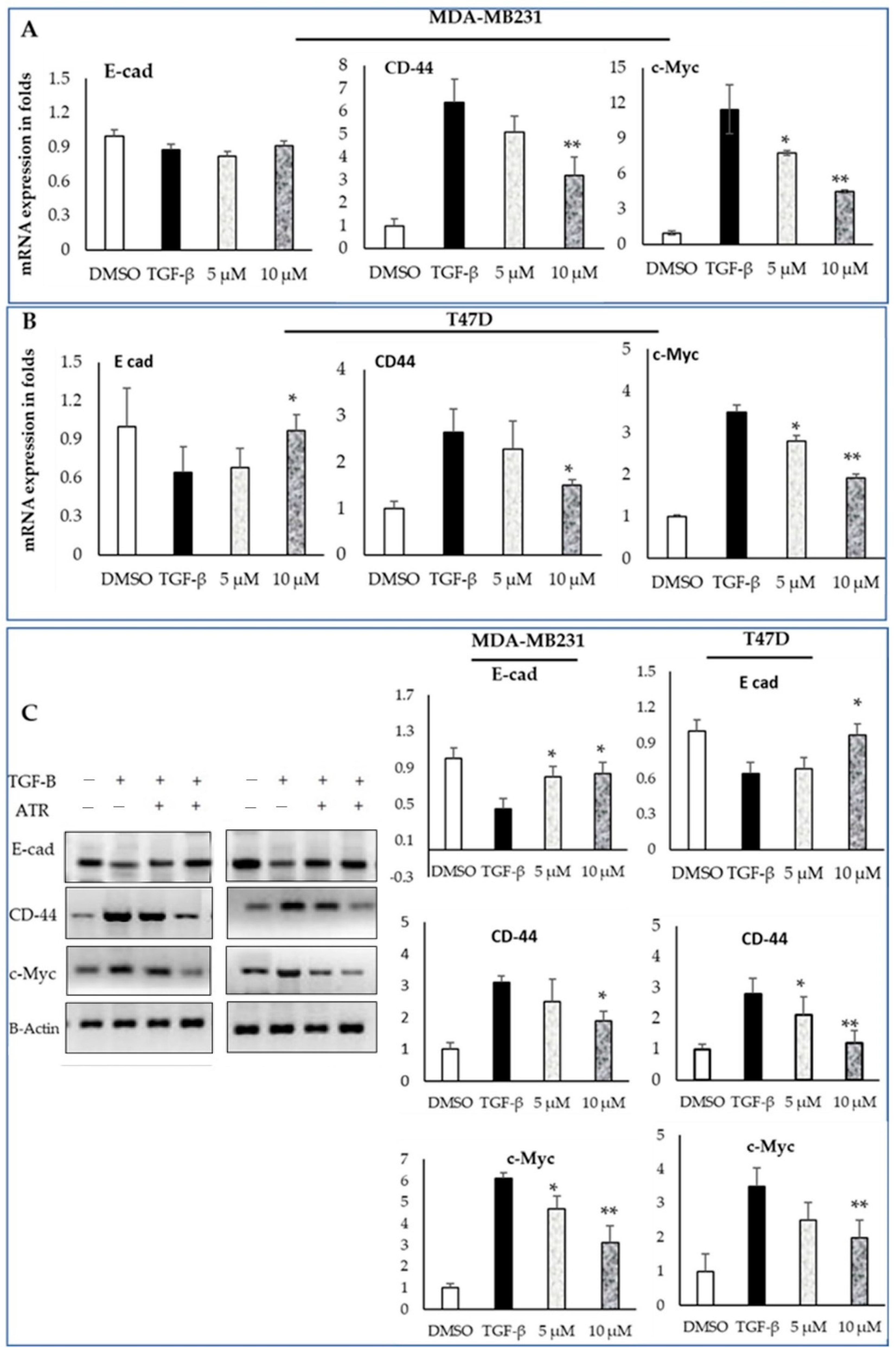
2.5. The Expression of Breast Cancer Mesenchymal Modification Genes Is Dysregulated after Atropine Treatment
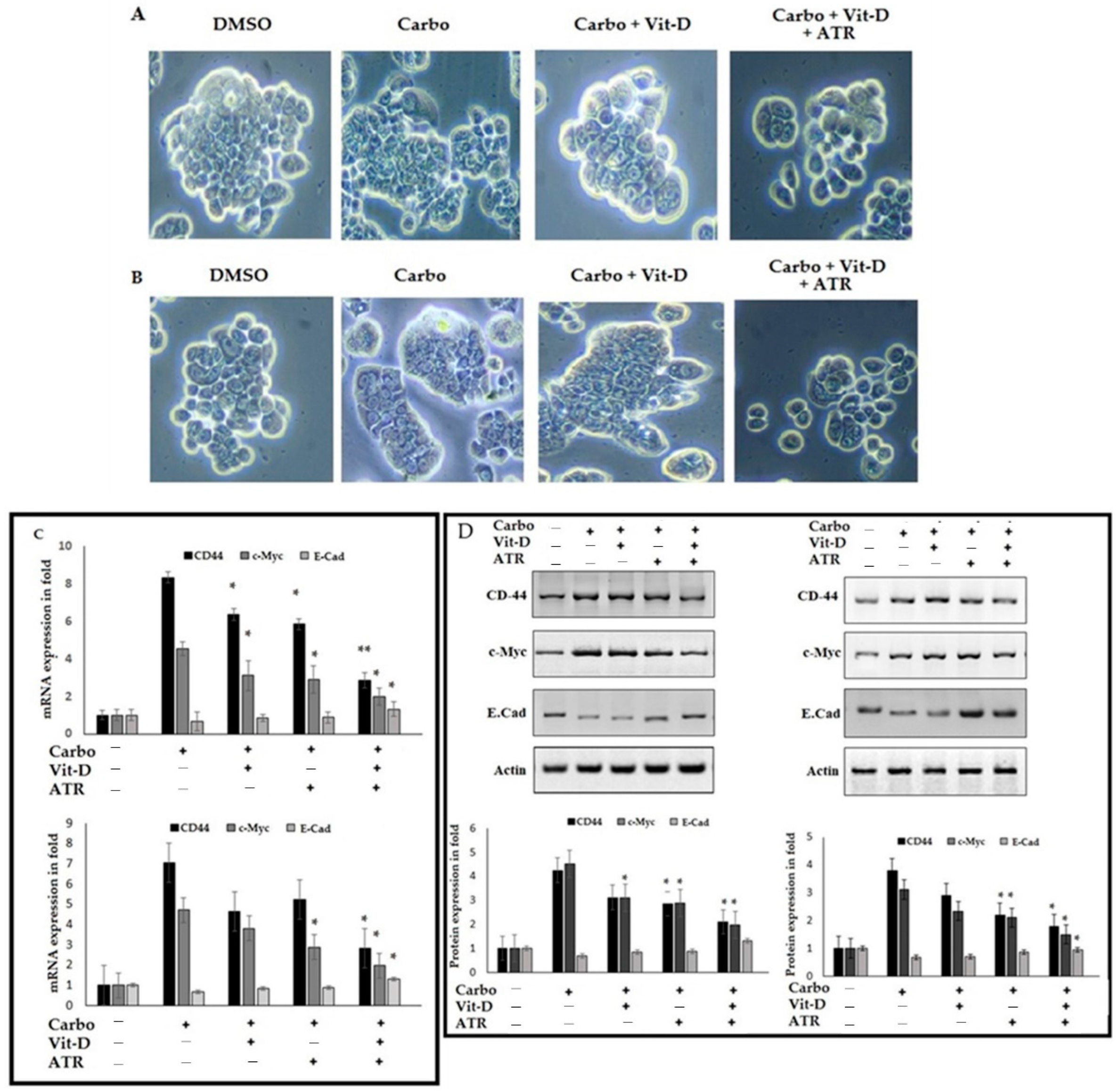
2.6. Atropine Abrogates the Carboplatin-Induced Cancer Stem Cell Modifications in Breast Cancer Cell Lines
2.7. Atropine Alleviates Carboplatin-Induced Mesenchymal Transition and Stemness in Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. In Silico Analysis
4.2. Cell Lines and Culture
4.3. Cell Proliferation and MTT Assay
4.4. Cell Migration Assay (Wound-Healing Assay)
4.5. Cell-Invasion Assay (Transwell Assay)
4.6. RNA Isolation and Quantitative Real-Time PCR
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- French, R.; Walter, G. Atropine. In Critical Care Toxicology; Springer: Greer, SC, USA, 2017; pp. 2725–2731. [Google Scholar]
- Espanol, A.J.; Jacob, G.; Dmytrenko, G.; Sales, M.E. Muscarinic activation enhances the anti-proliferative effect of paclitaxel in murine breast tumor cells. Anti-Cancer Agents Med. Chem. 2013, 13, 1273–1279. [Google Scholar] [CrossRef]
- Lombardi, M.G.; Negroni, M.P.; Pelegrina, L.T.; Castro, M.E.; Fiszman, G.L.; Azar, M.E.; Morgado, C.C.; Sales, M.E. Autoantibodies against muscarinic receptors in breast cancer: Their role in tumor angiogenesis. PLoS ONE 2013, 8, e57572. [Google Scholar]
- Salem, A.R.; Martinez Pulido, P.; Sanchez, F.; Sanchez, Y.; Espanol, A.J.; Sales, M.E. Effect of low dose metronomic therapy on MCF-7 tumor cells growth and angiogenesis. Role of muscarinic acetylcholine receptors. Int. Immunopharmacol. 2020, 84, 106514. [Google Scholar] [CrossRef]
- Chen, J.; Cheuk, I.W.; Shin, V.Y.; Kwong, A. Acetylcholine receptors: Key players in cancer development. Surg. Oncol. 2019, 31, 46–53. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Visvader, J.E.; Clevers, H. Tissue-specific designs of stem cell hierarchies. Nat. Cell Biol. 2016, 18, 349–355. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef]
- Mahdi, S.H.; Cheng, H.; Li, J.; Feng, R. The effect of TGF-beta-induced epithelial–mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells. Arch. Biochem. Biophys. 2015, 583, 18–26. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef]
- Jurrius, P.; Green, T.; Garmo, H.; Young, M.; Cariati, M.; Gillett, C.; Mera, A.; Harries, M.; Grigoriadis, A.; Pinder, S.; et al. Invasive breast cancer over four decades reveals persisting poor metastatic outcomes in treatment resistant subgroup—The “ATRESS” phenomenon. Breast 2020, 50, 39–48. [Google Scholar] [CrossRef]
- Spencer, A.; Sligar, A.D.; Chavarria, D.; Lee, J.; Choksi, D.; Patil, N.P.; Lee, H.; Veith, A.P.; Riley, W.J.; Desai, S.; et al. Biomechanical regulation of breast cancer metastasis and progression. Sci. Rep. 2021, 11, 9838. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Li, J.; Luco, A.L.; Camirand, A.; St-Arnaud, R.; Kremer, R. Vitamin D Regulates CXCL12/CXCR4 and Epithelial-to-Mesenchymal Transition in a Model of Breast Cancer Metastasis to Lung. Endocrinology 2021, 162, bqab049. [Google Scholar] [CrossRef]
- Negri, M.; Gentile, A.; de Angelis, C.; Monto, T.; Patalano, R.; Colao, A.; Pivonello, R.; Pivonello, C. Vitamin D-Induced Molecular Mechanisms to Potentiate Cancer Therapy and to Reverse Drug-Resistance in Cancer Cells. Nutrients 2020, 12, 1798. [Google Scholar] [CrossRef]
- Sales, M.E. Muscarinic Receptors as Targets for Metronomic Therapy in Breast Cancer. Curr. Pharm. Des. 2016, 22, 2170–2177. [Google Scholar] [CrossRef]
- Larriba, M.J.; de Herreros, A.G.; Muñoz, A. Vitamin D and the Epithelial to Mesenchymal Transition. Stem Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef]
- Feng, Y.; Hu, X.; Liu, G.; Lu, L.; Zhao, W.; Shen, F.; Ma, K.; Sun, C.; Zhu, C.; Zhang, B. M3 muscarinic acetylcholine receptors regulate epithelial–mesenchymal transition, perineural invasion, and migration/metastasis in cholangiocarcinoma through the AKT pathway. Cancer Cell Int. 2018, 18, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahia, M.; Benhouda, A.; Haba, H. New biological anticancer activities of atropine isolated from algerian hyoscyamus albus’s leaves. Pharmacologyonline 2018, 3, 286–296. [Google Scholar]
- Yang, K.; Song, Y.; Tang, Y.B.; Xu, Z.P.; Zhou, W.; Hou, L.N.; Zhu, L.; Yu, Z.H.; Chen, H.Z.; Cui, Y.Y. mAChRs activation induces epithelial–mesenchymal transition on lung epithelial cells. BMC Pulm. Med. 2014, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Sales, M.E.; Español, A.J.; Salem, A.R.; Pulido, P.M.; Sanchez, Y.; Sanchez, F. Role of Muscarinic Acetylcholine Receptors in Breast Cancer: Design of Metronomic Chemotherapy. Curr. Clin. Pharmacol. 2019, 14, 91–100. [Google Scholar] [CrossRef]
- Fiszman, G.L.; Middonno, M.C.; de la Torre, E.; Farina, M.; Español, A.J.; Sales, M.E. Activation of muscarinic cholinergic receptors induces MCF-7 cells proliferation and angiogenesis by stimulating nitric oxide synthase activity. Cancer Biol. Ther. 2007, 6, 1106–1113. [Google Scholar] [CrossRef]
- Rimmaudo, L.; de la Torre, E.; Sacerdote de Lustig, E.; Sales, M.E. mAChR are involved in murine mammary adenocOParcinoma cells LMM3 proliferation and angiogenesis. Biochem. Biophys. Res. Commun. 2005, 334, 1360–1365. [Google Scholar] [CrossRef]
- Horas, K.; Zheng, Y.; Fong-Yee, C.; Macfarlane, E.; Manibo, J.; Chen, Y.; Qiao, J.; Gao, M.; Haydar, N.; McDonald, M.M.; et al. Loss of the Vitamin D Receptor in Human Breast Cancer Cells Promotes Epithelial to Mesenchymal Cell Transition and Skeletal Colonization. J. Bone Miner. Res. 2019, 34, 1721–1732. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction with | Binding Energy | Ligand Efficiency | Intermole Energy | Ligand Atoms (Ring) | Docked Amino Acid Residue (Bond Length) |
|---|---|---|---|---|---|
| E-cad | −6.75 | −0.32 | −7.74 | Conventional hydrogen bond: C12-H Pi-Alkyl hydrophobic bond: O | Chain A: GLN’197’OE1’ (2.01 Å) Chain A: LEU’196’ (4.53 Å) |
| ZEB-2 | −4.78 | −0.23 | −5.85 | Conventional hydrogen bond: C12-H Carbon hydrogen bond interaction C8 C12-O Alkyl hydrophobic bond: O O Pi-Alkyl hydrophobic bond: O O | Chain A: VAL’46’O’(1.75 Å) Chain A: PRO’10’O’ (3.09 Å) Chain A: LYS’49’CE’ (3.72 Å) Chain A: LEU’15’ (5.38 Å) Chain A: LEU’15’ (5.26 Å) Chain A: LEU’15’ (5.38 Å) Chain A: LYS’49’’CE’ (3.72 Å) |
| Primer Name | Forward Primer | Reverse Primer | PCR Product Size |
|---|---|---|---|
| E-cad | GCCTCCTGAAAAGAGAGTGGAAG | TGGCAGTGTCTCTCCAAATCCG | 189 |
| CD-44 | CCAGAAGGAACAGTGGTTTGGC | ACTGTCCTCTGGGCTTGGTGTT | 212 |
| ZEB2 | AATGCACAGAGTGTGGCAAGGC | CTGCTGATGTGCGAACTGTAGG | 231 |
| c-MYC | CCTGGTGCTCCATGAGGAGAC | CAGACTCTGACCTTTTGCCAGG | 168 |
| SOX-2 | GCTACAGCATGATGCAGGACCA | TCTGCGAGCTGGTCATGGAGTT | 188 |
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA | 195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, E.A.; Alkuwayti, M.A.; Ibrahim, H.-I.M. Atropine Is a Suppressor of Epithelial–Mesenchymal Transition (EMT) That Reduces Stemness in Drug-Resistant Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9849. https://doi.org/10.3390/ijms23179849
Ahmed EA, Alkuwayti MA, Ibrahim H-IM. Atropine Is a Suppressor of Epithelial–Mesenchymal Transition (EMT) That Reduces Stemness in Drug-Resistant Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(17):9849. https://doi.org/10.3390/ijms23179849
Chicago/Turabian StyleAhmed, Emad A., Mayyadah A. Alkuwayti, and Hairul-Islam M. Ibrahim. 2022. "Atropine Is a Suppressor of Epithelial–Mesenchymal Transition (EMT) That Reduces Stemness in Drug-Resistant Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 17: 9849. https://doi.org/10.3390/ijms23179849
APA StyleAhmed, E. A., Alkuwayti, M. A., & Ibrahim, H.-I. M. (2022). Atropine Is a Suppressor of Epithelial–Mesenchymal Transition (EMT) That Reduces Stemness in Drug-Resistant Breast Cancer Cells. International Journal of Molecular Sciences, 23(17), 9849. https://doi.org/10.3390/ijms23179849