
Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model


Abstract
:1. Introduction
2. Results
2.1. The HCS1 Deficient Strain Is Not Hypersensitive to 4-Chlorophenol on Solid YPD Medium
2.2. The Deletion of HCS1 Leads to Hypersensitivity to Cycloheximide
2.3. Full-Length Human IGHMBP2 Suppresses the Hypersensitivity of hcs1Δ Mutant to Cycloheximide
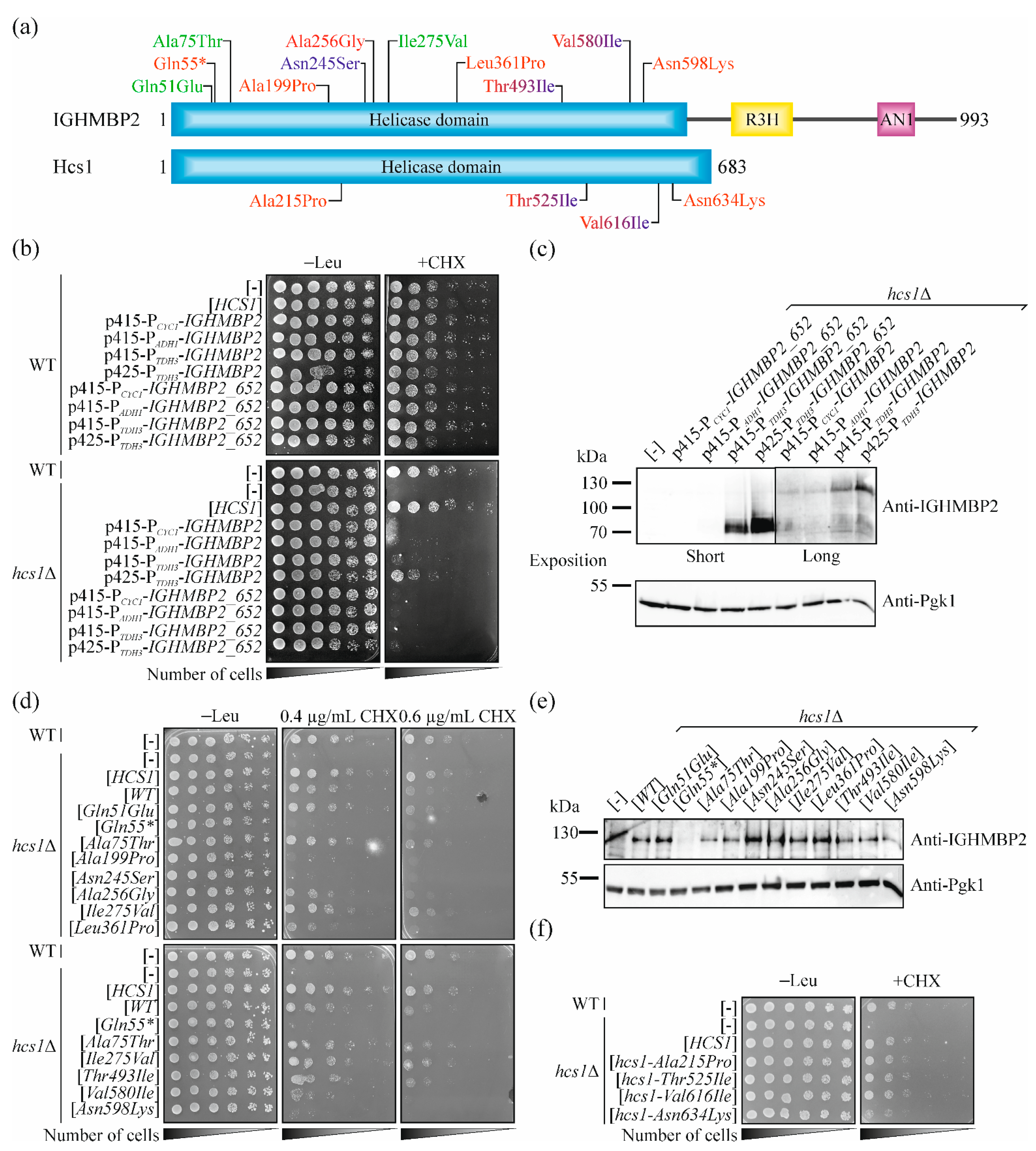
2.4. Selection of Missense Mutations in IGHMBP2 for Validation of Our Yeast System
2.5. Pathogenic Mutations in IGHMBP2 Gene but Not in HCS1 Results in a Loss-of-Function in Yeast Model
3. Discussion
4. Materials and Methods
4.1. Strains, Media, and Growth Conditions
4.2. Plasmids
4.3. Site-Directed Mutagenesis
4.4. Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jędrzejowska, M.; Madej-Pilarczyk, A.; Fidziańska, A.; Mierzewska, H.; Pronicka, E.; Obersztyn, E.; Gos, M.; Pronicki, M.; Kmieć, T.; Migdał, M.; et al. Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol. 2014, 18, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Saladini, M.; Nizzardo, M.; Govoni, A.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. Spinal muscular atrophy with respiratory distress type 1: Clinical phenotypes, molecular pathogenesis and therapeutic insights. J. Cell. Mol. Med. 2020, 24, 1169–1178. [Google Scholar] [CrossRef]
- Viguier, A.; Lauwers-Cances, V.; Cintas, P.; Manel, V.; Peudenier, S.; Desguerre, I.; Quijano-Roy, S.; Vanhulle, C.; Fradin, M.; Isapof, A.; et al. Spinal muscular atrophy with respiratory distress type 1: A multicenter retrospective study. Neuromuscul. Disord. 2019, 29, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Schuelke, M.; Bertini, E.; D’Amico, A.; Goemans, N.; Grohmann, K.; Hübner, C.; Varon, R. Genomic rearrangements at the IGHMBP2 gene locus in two patients with SMARD1. Hum. Genet. 2004, 115, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Handoko, L.; Varon, R.; Stephani, U.; Tsao, C.Y.; Mendell, J.R.; Lützkendorf, S.; Hübner, C.; von Au, K.; Jablonka, S.; et al. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): Determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J. Mol. Med. 2009, 87, 31–41. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Forrester, N.; Antoniadi, T.; Willis, T.; Sethuraman, S.K.; Samuels, M. Charcot Marie Tooth disease type 2S with late onset diaphragmatic weakness: An atypical case. Neuromuscul. Disord. 2018, 28, 1016–1021. [Google Scholar] [CrossRef]
- Tomaselli, P.J.; Horga, A.; Rossor, A.M.; Jaunmuktane, Z.; Cortese, A.; Blake, J.C.; Zarate-Lopez, N.; Houlden, H.; Reilly, M.M. IGHMBP2 mutation associated with organ-specific autonomic dysfunction. Neuromuscul. Disord. 2018, 28, 1012–1015. [Google Scholar] [CrossRef]
- Grohmann, K.; Schuelke, M.; Diers, A.; Hoffmann, K.; Lucke, B.; Adams, C.; Bertini, E.; Leonhardt-Horti, H.; Muntoni, F.; Ouvrier, R.; et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001, 29, 75–77. [Google Scholar] [CrossRef]
- Cottenie, E.; Kochanski, A.; Jordanova, A.; Bansagi, B.; Zimon, M.; Horga, A.; Jaunmuktane, Z.; Saveri, P.; Rasic, V.M.; Baets, J.; et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet. 2014, 95, 590–601. [Google Scholar] [CrossRef]
- Wagner, J.D.; Huang, L.; Tetreault, M.; Majewski, J.; Boycott, K.M.; Bulman, D.E.; Dyment, D.A.; McMillan, H.J.; Consortium, C.R.C. Autosomal recessive axonal polyneuropathy in a sibling pair due to a novel homozygous mutation in IGHMBP2. Neuromuscul. Disord. 2015, 25, 794–799. [Google Scholar] [CrossRef]
- Liu, L.; Li, X.; Hu, Z.; Mao, X.; Zi, X.; Xia, K.; Tang, B.; Zhang, R. IGHMBP2-related clinical and genetic features in a cohort of Chinese Charcot-Marie-Tooth disease type 2 patients. Neuromuscul. Disord. 2017, 27, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Berger, K.; Arafat, D.; Nallamilli, B.R.R.; Subramanian, H.P.; Joseph, S.; Anderson, M.E.; Campbell, K.P.; Glass, J.; Gibson, G.; et al. Clinical utility of RNA sequencing to resolve unusual GNE myopathy with a novel promoter deletion. Muscle Nerve 2019, 60, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Hegde, M. Clinical Utility of Transcriptome Sequencing: Toward a Better Diagnosis for Mendelian Disorders. Clin. Chem. 2018, 64, 882–884. [Google Scholar] [CrossRef]
- Molnar, G.M.; Crozat, A.; Kraeft, S.K.; Dou, Q.P.; Chen, L.B.; Pardee, A.B. Association of the mammalian helicase MAH with the pre-mRNA splicing complex. Proc. Natl. Acad. Sci. USA 1997, 94, 7831–7836. [Google Scholar] [CrossRef] [PubMed]
- Guenther, U.P.; Handoko, L.; Laggerbauer, B.; Jablonka, S.; Chari, A.; Alzheimer, M.; Ohmer, J.; Plöttner, O.; Gehring, N.; Sickmann, A.; et al. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1). Hum. Mol. Genet. 2009, 18, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Bowler, M.W.; Lai, T.F.; Song, H. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res. 2012, 40, 11009–11022. [Google Scholar] [CrossRef] [PubMed]
- Rzepnikowska, W.; Kochański, A. Models for IGHMBP2-associated diseases: An overview and a roadmap for the future. Neuromuscul. Disord. 2021, 31, 1266–1278. [Google Scholar] [CrossRef]
- Kerr, D.; Khalili, K. A recombinant cDNA derived from human brain encodes a DNA binding protein that stimulates transcription of the human neurotropic virus JCV. J. Biol. Chem. 1991, 266, 15876–15881. [Google Scholar] [CrossRef]
- Chen, N.N.; Kerr, D.; Chang, C.F.; Honjo, T.; Khalili, K. Evidence for regulation of transcription and replication of the human neurotropic virus JCV genome by the human S(mu)bp-2 protein in glial cells. Gene 1997, 185, 55–62. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.C.; Montalvo, E.A. Smubp-2 represses the Epstein-Barr virus lytic switch promoter. Virology 1999, 255, 160–170. [Google Scholar] [CrossRef] [Green Version]
- de Planell-Saguer, M.; Schroeder, D.G.; Rodicio, M.C.; Cox, G.A.; Mourelatos, Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet. 2009, 18, 2115–2126. [Google Scholar] [CrossRef]
- Surrey, V.; Zöller, C.; Lork, A.A.; Moradi, M.; Balk, S.; Dombert, B.; Saal-Bauernschubert, L.; Briese, M.; Appenzeller, S.; Fischer, U.; et al. Impaired Local Translation of β-actin mRNA in Ighmbp2-Deficient Motoneurons: Implications for Spinal Muscular Atrophy with respiratory Distress (SMARD1). Neuroscience 2018, 386, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Bonn, F.; Pantakani, K.; Shoukier, M.; Langer, T.; Mannan, A.U. Functional evaluation of paraplegin mutations by a yeast complementation assay. Hum. Mutat. 2010, 31, 617–621. [Google Scholar] [CrossRef]
- Franssens, V.; Bynens, T.; Van den Brande, J.; Vandermeeren, K.; Verduyckt, M.; Winderickx, J. The benefits of humanized yeast models to study Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 760629. [Google Scholar] [CrossRef] [PubMed]
- Verduyckt, M.; Vignaud, H.; Bynens, T.; Van den Brande, J.; Franssens, V.; Cullin, C.; Winderickx, J. Yeast as a Model for Alzheimer’s Disease: Latest Studies and Advanced Strategies. Methods Mol. Biol. 2016, 1303, 197–215. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef]
- Mason, R.P.; Giorgini, F. Modeling Huntington disease in yeast: Perspectives and future directions. Prion 2011, 5, 269–276. [Google Scholar] [CrossRef]
- Soczewka, P.; Kolakowski, D.; Smaczynska-de Rooij, I.; Rzepnikowska, W.; Ayscough, K.R.; Kaminska, J.; Zoladek, T. Yeast-model-based study identified myosin- and calcium-dependent calmodulin signalling as a potential target for drug intervention in chorea-acanthocytosis. Dis. Model. Mech. 2019, 12, dmm036830. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Binięda, K.; Kochański, A. A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: A Simple System for Complex, Heterogeneous Diseases. Int. J. Mol. Sci. 2020, 21, 865. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; di Punzio, G.; Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T.; Donnini, C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes 2021, 12, 300. [Google Scholar] [CrossRef]
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Zhou, D.; Lv, T.; Xu, A.; Wang, H.; Zhao, X.; Zhang, B.; Li, Y.; Jia, S.; et al. Complex ATP7B mutation patterns in Wilson disease and evaluation of a yeast model for functional analysis of variants. Hum. Mutat. 2019, 40, 552–565. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes 2020, 11, 310. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Flis, K.; Kaminska, J.; Grynberg, M.; Urbanek, A.; Ayscough, K.R.; Zoladek, T. Amino acid substitution equivalent to human chorea-acanthocytosis I2771R in yeast Vps13 protein affects its binding to phosphatidylinositol 3-phosphate. Hum. Mol. Genet. 2017, 26, 1497–1510. [Google Scholar] [CrossRef]
- Gilberti, M.; Baruffini, E.; Donnini, C.; Dallabona, C. Pathological alleles of MPV17 modeled in the yeast Saccharomyces cerevisiae orthologous gene SYM1 reveal their inability to take part in a high molecular weight complex. PLoS ONE 2018, 13, e0205014. [Google Scholar] [CrossRef]
- Kolakowski, D.; Rzepnikowska, W.; Kaniak-Golik, A.; Zoladek, T.; Kaminska, J. The GTPase Arf1 Is a Determinant of Yeast Vps13 Localization to the Golgi Apparatus. Int. J. Mol. Sci. 2021, 22, 12274. [Google Scholar] [CrossRef]
- Soczewka, P.; Flis, K.; Tribouillard-Tanvier, D.; di Rago, J.P.; Santos, C.N.; Menezes, R.; Kaminska, J.; Zoladek, T. Flavonoids as Potential Drugs for VPS13-Dependent Rare Neurodegenerative Diseases. Genes 2020, 11, 828. [Google Scholar] [CrossRef]
- Binięda, K.; Rzepnikowska, W.; Kolakowski, D.; Kaminska, J.; Szczepankiewicz, A.A.; Nieznańska, H.; Kochański, A.; Kabzińska, D. Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network. Int. J. Mol. Sci. 2021, 22, 914. [Google Scholar] [CrossRef]
- Yadav, V.; Shitiz, K.; Pandey, R.; Yadav, J. Chlorophenol stress affects aromatic amino acid biosynthesis—A genome-wide study. Yeast 2011, 28, 81–91. [Google Scholar] [CrossRef]
- Collart, M.A. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2016, 7, 438–454. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, L.; Jiang, H. Doa1 targets ubiquitinated substrates for mitochondria-associated degradation. J. Cell Biol. 2016, 213, 49–63. [Google Scholar] [CrossRef]
- Pluta, K.; Lefebvre, O.; Martin, N.C.; Smagowicz, W.J.; Stanford, D.R.; Ellis, S.R.; Hopper, A.K.; Sentenac, A.; Boguta, M. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 5031–5040. [Google Scholar] [CrossRef]
- Kunz, J.; Loeschmann, A.; Deuter-Reinhard, M.; Hall, M.N. FAP1, a homologue of human transcription factor NF-X1, competes with rapamycin for binding to FKBP12 in yeast. Mol. Microbiol. 2000, 37, 1480–1493. [Google Scholar] [CrossRef]
- Cieśla, M.; Makała, E.; Płonka, M.; Bazan, R.; Gewartowski, K.; Dziembowski, A.; Boguta, M. Rbs1, a new protein implicated in RNA polymerase III biogenesis in yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 2015, 35, 1169–1181. [Google Scholar] [CrossRef]
- Lebaron, S.; Papin, C.; Capeyrou, R.; Chen, Y.L.; Froment, C.; Monsarrat, B.; Caizergues-Ferrer, M.; Grigoriev, M.; Henry, Y. The ATPase and helicase activities of Prp43p are stimulated by the G-patch protein Pfa1p during yeast ribosome biogenesis. EMBO J. 2009, 28, 3808–3819. [Google Scholar] [CrossRef]
- Kanaan, J.; Raj, S.; Decourty, L.; Saveanu, C.; Croquette, V.; Le Hir, H. UPF1-like helicase grip on nucleic acids dictates processivity. Nat. Commun. 2018, 9, 3752. [Google Scholar] [CrossRef]
- Guenther, U.P.; Varon, R.; Schlicke, M.; Dutrannoy, V.; Volk, A.; Hübner, C.; von Au, K.; Schuelke, M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): Defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat. 2007, 28, 808–815. [Google Scholar] [CrossRef]
- Eckart, M.; Guenther, U.P.; Idkowiak, J.; Varon, R.; Grolle, B.; Boffi, P.; Van Maldergem, L.; Hübner, C.; Schuelke, M.; von Au, K. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics 2012, 129, e148–e156. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef]
- Grohmann, K.; Varon, R.; Stolz, P.; Schuelke, M.; Janetzki, C.; Bertini, E.; Bushby, K.; Muntoni, F.; Ouvrier, R.; Van Maldergem, L.; et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol. 2003, 54, 719–724. [Google Scholar] [CrossRef]
- Pitt, M.; Houlden, H.; Jacobs, J.; Mok, Q.; Harding, B.; Reilly, M.; Surtees, R. Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain 2003, 126, 2682–2692. [Google Scholar] [CrossRef]
- Pierson, T.M.; Tart, G.; Adams, D.; Toro, C.; Golas, G.; Tifft, C.; Gahl, W. Infantile-onset spinal muscular atrophy with respiratory distress-1 diagnosed in a 20-year-old man. Neuromuscul. Disord. 2011, 21, 353–355. [Google Scholar] [CrossRef]
- Stalpers, X.L.; Verrips, A.; Poll-The, B.T.; Cobben, J.M.; Snoeck, I.N.; de Coo, I.F.; Brooks, A.; Bulk, S.; Gooskens, R.; Fock, A.; et al. Clinical and mutational characteristics of spinal muscular atrophy with respiratory distress type 1 in The Netherlands. Neuromuscul. Disord. 2013, 23, 461–468. [Google Scholar] [CrossRef]
- Joseph, S.; Robb, S.A.; Mohammed, S.; Lillis, S.; Simonds, A.; Manzur, A.Y.; Walter, S.; Wraige, E. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul. Disord. 2009, 19, 193–195. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Longman, C.; O’Hara, A.; Kirkpatrick, M.; McWilliam, R. Growing up with spinal muscular atrophy with respiratory distress (SMARD1). Neuromuscul. Disord. 2015, 25, 169–171. [Google Scholar] [CrossRef]
- Pedurupillay, C.R.; Amundsen, S.S.; Barøy, T.; Rasmussen, M.; Blomhoff, A.; Stadheim, B.F.; Ørstavik, K.; Holmgren, A.; Iqbal, T.; Frengen, E.; et al. Clinical and molecular characteristics in three families with biallelic mutations in IGHMBP2. Neuromuscul. Disord. 2016, 26, 570–575. [Google Scholar] [CrossRef]
- Wong, V.C.; Chung, B.H.; Li, S.; Goh, W.; Lee, S.L. Mutation of gene in spinal muscular atrophy respiratory distress type I. Pediatr. Neurol. 2006, 34, 474–477. [Google Scholar] [CrossRef]
- Bansagi, B.; Griffin, H.; Whittaker, R.G.; Antoniadi, T.; Evangelista, T.; Miller, J.; Greenslade, M.; Forester, N.; Duff, J.; Bradshaw, A.; et al. Genetic heterogeneity of motor neuropathies. Neurology 2017, 88, 1226–1234. [Google Scholar] [CrossRef]
- Cox, G.A.; Mahaffey, C.L.; Frankel, W.N. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron 1998, 21, 1327–1337. [Google Scholar] [CrossRef]
- Smith, C.E.; Lorson, M.A.; Ricardez Hernandez, S.M.; Al Rawi, Z.; Mao, J.; Marquez, J.; Villalón, E.; Keilholz, A.N.; Smith, C.L.; Garro-Kacher, M.O.; et al. The Ighmbp2D564N mouse model is the first SMARD1 model to demonstrate respiratory defects. Hum. Mol. Genet. 2022, 31, 1293–1307. [Google Scholar] [CrossRef]
- Karkusiewicz, I.; Turowski, T.W.; Graczyk, D.; Towpik, J.; Dhungel, N.; Hopper, A.K.; Boguta, M. Maf1 protein, repressor of RNA polymerase III, indirectly affects tRNA processing. J. Biol. Chem. 2011, 286, 39478–39488. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 1989, 122, 19–27. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| IGHMBP2 Variant | ClinVar Status | Clinical Phenotype | In Vitro Phenotypes | Functionality in CHX-Hypersensitivity Test | Phenotype Compatibility | Comments |
|---|---|---|---|---|---|---|
| c.151C > G p.Gln51Glu | Benign/Likely benign/Uncertain significance | n.a. | n.t. | Functional protein | Good | |
| c.163C > T p.Gln55* | Pathogenic | SMARD1 | n.t. | Non-functional protein | Good | Compound heterozygosity p.Gln55*/p.Gln657* [47] |
| c.223G > A p.Ala75Thr | Benign | n.a. | n.t. | Functional protein | Good | |
| c.595G > A p.Ala199Pro | Uncertain significance | SMARD1 | n.t. | Non-functional protein | Good | Compound heterozygosity p.Ala199Pro/p.Ser539_Tyr541del [1] |
| c.734A > G p.Asn245Ser | Uncertain significance | CMT2S; Independent ambulation | n.t. | Non-functional protein | Medium | Compound heterozygosity, but second allele was not identified [9] |
| c.767C > G p.Al256Gly | Uncertain significance | SMARD1 | n.t. | Partially functional protein | Good | In patient, three different mutations in IGHMBP2 were identified and confirmed (p.Ala256Gly, p.Ala398Glu and p.Glu514Lys) [49]; also in heterozygous state [58] |
| c.823A > G p.Ile275Val | Benign | n.a. | n.t. | Functional protein | Good | |
| c.1082T > C p.Leu361Pro | Pathogenic | Infantile SMARD1 (with pCys496*; p.Leu577Pro; c.1060 + 1G > T; p.GLu382Lys or p.Glu514Lys); juvenile SMARD1 (with p.Thr493Ile) | No ATPase or helicase activity [15] | Partially functional protein | Good | Compound heterozygosity: p.Leu361Pro/p.Cys496* [51] p.Leu361Pro/p.Leu577Pro [50]; p.Leu361Pro/c.1060 + 1G > T [47] p.Leu361Pro/ p.Thr493Ile [5]; p.Leu361Pro/ p.Glu382Lys [52]; p.Leu361Pro/p.Glu514Lys [53] |
| c.1478C > T p.Thr493Ile | Pathogenic/Likely pathogenic | Infantile SMARD1 (with p.Arg788*; p.Cys496*; p.Leu155Gln; p.Glu514Lys or c.86 + 1022_c.257–191del2894); Juvenile SMARD1 (with p.Leu361Pro); CMT2S (with p.Lys328Thrfs46* but SMARD1 in patient with Kabuki syndrome) | ATPase and helicase activity as in WT; decrease in RNA/DNA binding capacity; tendency to form aggregates and to degrade [5,15] | Functional protein | Medium | Compound heterozygosity: p.Leu361Pro/p.Thr493Ile; pThr493Ile/p.Arg788* [5] p.Thr493Ile/p.Cys496* [54] p.Leu155Gln/p.Thr493Ile [55] p.Thr493Ile/p.Lys328Thrfs46* [56]; p.Thr493Ile/ p.Glu514Lys p.Thr493Ile/c.86 + 1022_c.257–191del2894 [53] |
| c.1738G > A p.Val580Ile | Likely pathogenic | SMARD1 ((homozygotes and p.Arg785fs; CMT2S (with Pro531Thr) | No ATPase or helicase activity [48] | Partially functional protein | Good | In homozygous state [8,48] and compound heterozygosity: p.Val580Ile/p.Pro531Thr [9]; p.Val580Ile/p.Arg785fs [57] |
| c.1794C > A p.Asn598Lys | Uncertain significance | SMARD1 | n.t. | Non-functional protein | Good | Compound heterozygosity: p.Arg147*/p.Asn598Lys [1] |
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Open Biosystem |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Open Biosystem |
| ccr4Δ | BY4741 ccr4::KanMX | This study |
| doa1Δ | BY4741 doa1::KanMX | Open Biosystem |
| fap1Δ | BY4741 fap1::KanMX | Open Biosystem |
| maf1Δ | BY4742 maf1::KanMX | [61] |
| rbs1Δ | BY4741 rbs1::KanMX | [44] |
| sqs1Δ | BY4741 sqs1::KanMX | Open Biosystem |
| hcs1Δ1 | BY4741 hcs1::NatMX | This study |
| hcs1Δ2 | BY4742 hcs1::NatMX | This study |
| ccr4Δ hcs1Δ | BY4741 ccr4::KanMX hcs1::NatMX | This study |
| doa1Δ hcs1Δ | BY4741 doa1::KanMX hcs1::NatMX | This study |
| fap1Δ hcs1Δ | BY4741 fap1::KanMX hcs1::NatMX | This study |
| maf1Δ hcs1Δ | BY4742 maf1::KanMX hcs1::NatMX | This study |
| rbs1Δ hcs1Δ | BY4741 rbs1::KanMX hcs1::NatMX | This study |
| sqs1Δ hcs1Δ | BY4741 sqs1::KanMX hcs1::NatMX | This study |
| Plasmid | Source |
|---|---|
| pRS315 [CEN6 LEU2] | [62] |
| pRS416 [CEN6 URA3] | [62] |
| p415-PCYC1 [CEN6 LEU2] | [63] |
| p415-PADH1 [CEN6 LEU2] | [63] |
| p415-PTDH3 [CEN6 LEU2] | [63] |
| p425-PTDH3 [2µ LEU2] | [63] |
| pRS315-HCS1 | This study |
| pRS416-HCS1 | This study |
| pRS315-HCS1-Ala215Pro | This study |
| pRS315-HCS1-Thr525Ile | This study |
| pRS315-HCS1-Val616Ile | This study |
| pRS315-HCS1-Asn634Lys | This study |
| pCMV3-IGHMBP2 | Sino Biological |
| p415-PCYC1-IGHMBP2_652 | This study |
| p415-PADH1-IGHMBP2_652 | This study |
| p415-PTDH3-IGHMBP2_652 | This study |
| p425-PTDH3-IGHMBP2_652 | This study |
| p415-PCYC1-IGHMBP2 | This study |
| p415-PADH1-IGHMBP2 | This study |
| p415-PTDH3-IGHMBP2 | This study |
| p425-PTDH3-IGHMBP2 | This study |
| p425-PTDH3-IGHMBP2-Gln51Glu | This study |
| p425-PTDH3-IGHMBP2-Gln55* | This study |
| p425-PTDH3-IGHMBP2-Ala75Thr | This study |
| p425-PTDH3-IGHMBP2-Ala199Pro | This study |
| p425-PTDH3-IGHMBP2-Asn245Ser | This study |
| p425-PTDH3-IGHMBP2-Al256Gly | This study |
| p425-PTDH3-IGHMBP2-Ile275Val | This study |
| p425-PTDH3-IGHMBP2-Leu361Pro | This study |
| p425-PTDH3-IGHMBP2-Thr493Ile | This study |
| p425-PTDH3-IGHMBP2-Val580Ile | This study |
| p425-PTDH3-IGHMBP2-Asn598Lys | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzepnikowska, W.; Kaminska, J.; Kochański, A. Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model. Int. J. Mol. Sci. 2022, 23, 9913. https://doi.org/10.3390/ijms23179913
Rzepnikowska W, Kaminska J, Kochański A. Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model. International Journal of Molecular Sciences. 2022; 23(17):9913. https://doi.org/10.3390/ijms23179913
Chicago/Turabian StyleRzepnikowska, Weronika, Joanna Kaminska, and Andrzej Kochański. 2022. "Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model" International Journal of Molecular Sciences 23, no. 17: 9913. https://doi.org/10.3390/ijms23179913
APA StyleRzepnikowska, W., Kaminska, J., & Kochański, A. (2022). Validation of the Pathogenic Effect of IGHMBP2 Gene Mutations Based on Yeast S. cerevisiae Model. International Journal of Molecular Sciences, 23(17), 9913. https://doi.org/10.3390/ijms23179913