
Strategies for Therapeutic Amelioration of Aberrant Plasma Zn2+ Handling in Thrombotic Disease: Targeting Fatty Acid/Serum Albumin-Mediated Effects

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
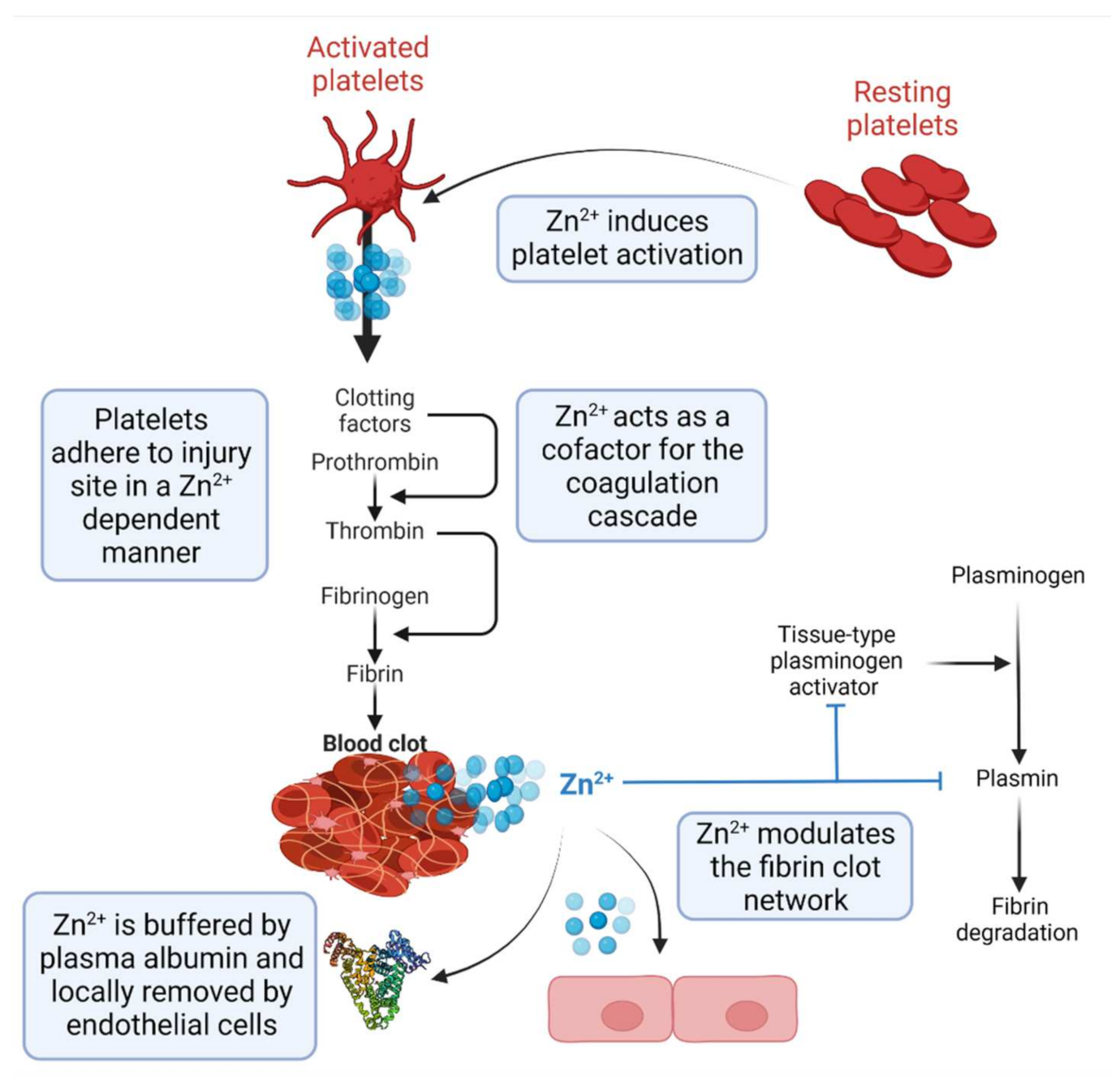
2. Zn2+ as a Regulator of Haemostasis
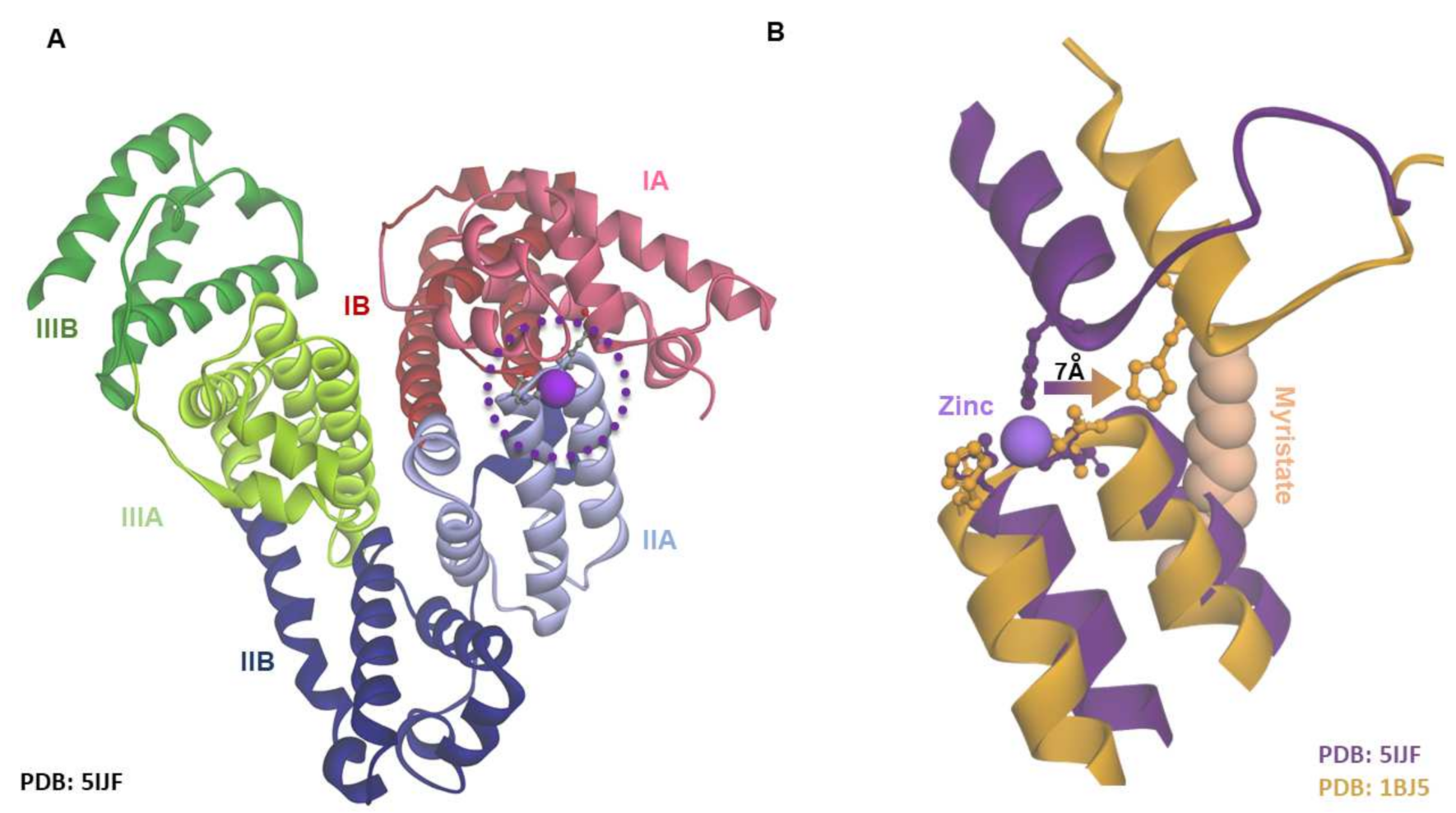
3. Zn2+ Binding by Human Serum Albumin and Effects of Non-Esterified Fatty Acids
4. Interventions to Ameliorate NEFA-Induced Alteration in Plasma Zn2+-Handling
4.1. Non-Pharmaceutical Interventions
4.1.1. Exercise
4.1.2. Dietary Changes
4.1.3. Omega-n NEFA Supplementation
4.1.4. Bariatric Surgery
5. Drug-Based Interventions
5.1. Statins
5.2. Fibrates
5.3. GLP-1 Receptor Agonists
5.4. Fatty Acid Synthase Inhibitors
5.5. Lipase Inhibitors
5.6. Cholesteryl Ester Transfer Protein Inhibitors (CETP Inhibitors)
6. Zn2+-Targeted Interventions
6.1. Chelation Agents
6.2. Targeting the FA2 Site of HSA
6.3. Downstream Targets of Disrupted Zinc Buffering
7. Conclusions
Funding
Conflicts of Interest
References
- Baumgartner, H.R.; Haudenschild, C. Adhesion of platelets to subendothelium. Ann. N. Y. Acad. Sci. 1972, 201, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Beumer, S.; Heijnen, H.; IJsseldijk, M.; Orlando, E.; de Groot, P.; Sixma, J. Platelet adhesion to fibronectin in flow: The importance of von Willebrand factor and glycoprotein Ib. Blood 1995, 86, 3452–3460. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Renné, T.; Schmaier, A.H.; Nickel, K.F.; Blombäck, M.; Maas, C. In vivo roles of factor XII. Blood 2012, 120, 4296–4303. [Google Scholar] [CrossRef]
- Hierons, S.J.; Marsh, J.S.; Wu, D.; Blindauer, C.A.; Stewart, A.J. The interplay between non-esterified fatty acids and plasma zinc and its influence on thrombotic risk in obesity and type 2 diabetes. Int. J. Mol. Sci. 2021, 22, 10140. [Google Scholar] [CrossRef] [PubMed]
- Nieuwdorp, M.; Stroes, E.S.; Meijers, J.C.; Büller, H. Hypercoagulability in the metabolic syndrome. Curr. Opin. Pharmacol. 2005, 5, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A. Vascular complications of diabetes. Circ. Res. 2016, 118, 1771–1785. [Google Scholar] [CrossRef]
- Blokhin, I.O.; Lentz, S.R. Mechanisms of thrombosis in obesity. Curr. Opin. Hematol. 2013, 20, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 8. [Google Scholar] [CrossRef]
- Maino, A.; Rosendaal, F.R.; Algra, A.; Peyvandi, F.; Siegerink, B. Hypercoagulability is a stronger risk factor for ischaemic stroke than for myocardial infarction: A systematic review. PLoS One 2015, 10, e0133523. [Google Scholar]
- Jackson, S.P. Arterial thrombosis—Insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: Pathogenesis, diagnosis, and medical management. Cardiovasc. Diagn. Ther. 2017, 7, S276–S284. [Google Scholar] [CrossRef]
- Carlsson, M.; Wessman, Y.; Almgren, P.; Groop, L. High levels of nonesterified fatty acids are associated with increased familial risk of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1588–1594. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Katundu, K.G.H.; Phoenix, F.A.; Khazaipoul, S.; Yu, R.; Lampiao, F.; Stefanowicz, F.; Blindauer, C.A.; Pitt, S.J.; Smith, T.K.; et al. Albumin-mediated alteration of plasma zinc speciation by fatty acids modulates blood clotting in type-2 diabetes. Chem. Sci. 2021, 12, 4079–4093. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Braun, A. Zinc homeostasis in platelet-related diseases. Int. J. Mol. Sci. 2019, 20, 5258. [Google Scholar] [CrossRef]
- Taylor, K.A.; Pugh, N. The contribution of zinc to platelet behaviour during haemostasis and thrombosis. Metallomics 2016, 8, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Fredenburgh, J.; Weitz, J. Zinc: An important cofactor in haemostasis and thrombosis. Thromb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef]
- Watson, B.R.; White, N.A.; Taylor, K.A.; Howes, J.-M.; Malcor, J.-D.M.; Bihan, D.; Sage, S.O.; Farndale, R.W.; Pugh, N. Zinc is a transmembrane agonist that induces platelet activation in a tyrosine phosphorylation-dependent manner. Metallomics 2016, 8, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Influence of zinc on glycosaminoglycan neutralisation during coagulation. Metallomics 2018, 10, 1180–1190. [Google Scholar] [CrossRef]
- Mahdi, F.; Madar, Z.S.; Figueroa, C.D.; Schmaier, A.H. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, GC1qR, and cytokeratin 1 on endothelial cell membranes. Blood 2002, 99, 3585–3596. [Google Scholar] [CrossRef] [PubMed]
- Gorodetsky, R.; Mou, X.; Blankenfeld, A.; Marx, G. Platelet multielemental composition, lability, and subcellular localization. Am. J. Hematol. 1993, 42, 278–283. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Glycosaminoglycan neutralization in coagulation control. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1258–1270. [Google Scholar] [CrossRef]
- Borza, D.-B.; Morgan, W.T. Histidine-proline-rich glycoprotein as a plasma pH sensor: Modulation of its interaction with glycosaminoglycans by pH and metals. J. Biol. Chem. 1998, 273, 5493–5499. [Google Scholar] [CrossRef] [PubMed]
- Kassaar, O.; Schwarz-Linek, U.; Blindauer, C.A.; Stewart, A.J. Plasma free fatty acid levels influence Zn2+-dependent histidine-rich glycoprotein–heparin interactions via an allosteric switch on serum albumin. J. Thromb. Haemost. 2015, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Hulett, M.D.; Parish, C.R. Histidine-rich glycoprotein binds to cell-surface heparan sulfate via its N-terminal domain following Zn2+ chelation. J. Biol. Chem. 2004, 279, 30114–30122. [Google Scholar] [CrossRef] [PubMed]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal structure of human fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef]
- Fredenburgh, J.C.; Leslie, B.A.; Stafford, A.R.; Lim, T.; Chan, H.H.; Weitz, J.I. Zn2+ mediates high affinity binding of heparin to the αc domain of fibrinogen. J. Biol. Chem. 2013, 288, 29394–29402. [Google Scholar] [CrossRef]
- Shibayama, Y.; Brunnee, T.; Kaplan, A.P.; Reddigari, S. Activation of human hageman factor (factor xii) in the presence of zinc and phosphate ions. Braz. J. Med. Biol. Res. 1994, 27, 1817–1828. [Google Scholar]
- Bernardo, M.M.; Day, D.E.; Halvorson, H.R.; Olson, S.T.; Shore, J.D. Surface-independent acceleration of factor xii activation by zinc ions. ii. direct binding and fluorescence studies. J. Biol. Chem. 1993, 268, 12477–12483. [Google Scholar] [CrossRef]
- Meier, H.L.; Pierce, J.V.; Colman, R.W.; Kaplan, A.P. Activation and function of human hageman factor. the role of high molecular weight kininogen and prekallikrein. J. Clin. Investig. 1977, 60, 18–31. [Google Scholar] [CrossRef]
- Baglia, F.A.; Gailani, D.; López, J.A.; Walsh, P.N. Identification of a binding site for glycoprotein ibα in the apple 3 domain of factor XI. J. Biol. Chem. 2004, 279, 45470–45476. [Google Scholar] [CrossRef]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscl. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Petersen, L.C.; Olsen, O.H.; Nielsen, L.S.; Freskgård, P.-O.; Persson, E. Binding of Zn2+ to a Ca2+ loop allosterically attenuates the activity of factor VIIa and reduces its affinity for tissue factor. Protein Sci. 2000, 9, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Marx, G. Zinc binding to fibrinogen and fibrin. Arch. Biochem. Biophys. 1988, 266, 285–288. [Google Scholar] [CrossRef]
- Butkowski, R.; King, D.; Cortes, P. Zinc(II) chelate binds to fibrinogen and its αC region. Adv. Biochem. 2021, 9, 11. [Google Scholar] [CrossRef]
- Henderson, S.J.; Xia, J.; Wu, H.; Stafford, A.R.; Leslie, B.A.; Fredenburgh, J.C.; Weitz, D.A.; Weitz, J.I. Zinc promotes clot stability by accelerating clot formation and modifying fibrin structure. Thromb. Haemost. 2016, 115, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Marx, P.F.; Bouma, B.N.; Meijers, J.C.M. Role of zinc ions in activation and inactivation of thrombin-activatable fibrinolysis inhibitor. Biochemistry 2002, 41, 1211–1216. [Google Scholar] [CrossRef]
- Hotz, C.; Peerson, J.M.; Brown, K.H. Suggested lower cutoffs of serum zinc concentrations for assessing zinc status: Reanalysis of the second national health and nutrition examination survey data (1976–1980). Am. J. Clin. Nutr. 2003, 78, 756–764. [Google Scholar] [CrossRef]
- Handing, K.B.; Shabalin, I.G.; Kassaar, O.; Khazaipoul, S.; Blindauer, C.A.; Stewart, A.J.; Chruszcz, M.; Minor, W. Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chem. Sci. 2016, 7, 6635–6648. [Google Scholar] [CrossRef]
- van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 7. [Google Scholar] [CrossRef]
- Blindauer, C.A.; Harvey, I.; Bunyan, K.E.; Stewart, A.J.; Sleep, D.; Harrison, D.J.; Berezenko, S.; Sadler, P.J. Structure, properties, and engineering of the major zinc binding site on human albumin. J. Biol. Chem. 2009, 284, 23116–23124. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.J.; Blindauer, C.A.; Berezenko, S.; Sleep, D.; Sadler, P.J. Interdomain zinc site on human albumin. Proc. Natl. Acad. Sci. USA 2003, 100, 3701–3706. [Google Scholar] [CrossRef] [PubMed]
- Bal, W.; Sokołowska, M.; Kurowska, E.; Faller, P. Binding of transition metal ions to albumin: Sites, affinities and rates. Biochim. Biophys. Acta 2013, 1830, 5444–5455. [Google Scholar] [CrossRef] [PubMed]
- Jouven, X.; Charles, M.-A.; Desnos, M.; Ducimetière, P. Circulating nonesterified fatty acid level as a predictive risk factor for sudden death in the population. Circulation 2001, 104, 756–761. [Google Scholar] [CrossRef]
- Petitpas, I.; Grüne, T.; Bhattacharya, A.A.; Curry, S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J. Mol. Biol. 2001, 314, 955–960. [Google Scholar] [CrossRef]
- Barnett, J.P.; Blindauer, C.A.; Kassaar, O.; Khazaipoul, S.; Martin, E.M.; Sadler, P.J.; Stewart, A.J. Allosteric modulation of zinc speciation by fatty acids. Biochim. Biophys. Acta 2013, 1830, 5456–5464. [Google Scholar] [CrossRef]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Mol. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Jacobs, D.R., Jr.; Steinberger, J.; Moran, A.; Steffen, L.M.; Sinaiko, A.R. Relation between serum free fatty acids and adiposity, insulin resistance, and cardiovascular risk factors from adolescence to adulthood. Diabetes 2013, 62, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.-K.; Jung, J.-M.; Kim, J.H.; Koh, S.-B.; Bang, O.-Y.; Oh, K. Free fatty acid is associated with thrombogenicity in cardioembolic stroke. Cerebrovasc. Dis. 2017, 44, 160–168. [Google Scholar] [CrossRef]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Frayn, K.N. Fat as a fuel: Emerging understanding of the adipose tissue-skeletal muscle axis: Adipose tissue-muscle interaction. Acta Physiol. 2010, 199, 509–518. [Google Scholar] [CrossRef]
- Weiss, C.; Seitel, G.; Bärtsch, P. Coagulation and fibrinolysis after moderate and very heavy exercise in healthy male subjects. Med. Sci. Sports Exerc. 1998, 30, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Shojaee-Moradie, F.; Baynes, K.C.R.; Pentecost, C.; Bell, J.D.; Thomas, E.L.; Jackson, N.C.; Stolinski, M.; Whyte, M.; Lovell, D.; Bowes, S.B.; et al. Exercise training reduces fatty acid availability and improves the insulin sensitivity of glucose metabolism. Diabetologia 2007, 50, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.; Holdaway, C.; Varma, T.; Petocz, P.; Samman, S. Lower serum zinc concentration despite higher dietary zinc intake in athletes: A systematic review and meta-analysis. Sports Med. 2018, 48, 327–336. [Google Scholar] [CrossRef] [PubMed]
- DeSouza, C.A.; Jones, P.P.; Seals, D.R. Physical activity status and adverse age-related differences in coagulation and fibrinolytic factors in women. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 362–368. [Google Scholar] [CrossRef]
- Stratton, J.R.; Chandler, W.L.; Schwartz, R.S.; Cerqueira, M.D.; Levy, W.C.; Kahn, S.E.; Larson, V.G.; Cain, K.C.; Beard, J.C.; Abrass, I.B. Effects of physical conditioning on fibrinolytic variables and fibrinogen in young and old healthy adults. Circulation 1991, 83, 1692–1697. [Google Scholar] [CrossRef]
- Wang, J.-S. Exercise prescription and thrombogenesis. J. Biomed. Sci. 2006, 13, 753–761. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Davis, C.; Bryan, J.; Hodgson, J.; Murphy, K. Definition of the mediterranean diet; A literature review. Nutrients 2015, 7, 9139–9153. [Google Scholar] [CrossRef] [PubMed]
- Lairon, D. Intervention studies on mediterranean diet and cardiovascular risk. Mol. Nutr. Food Res. 2007, 51, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, T.; Ninomiya, T.; Wang, A.; Neal, B.; Jun, M.; Wong, M.G.; Jardine, M.; Hillis, G.S.; Perkovic, V. Effects of the Mediterranean diet on cardiovascular outcomes—A systematic review and meta-analysis. PLoS One 2016, 11, e0159252. [Google Scholar] [CrossRef]
- Hernáez, Á.; Castañer, O.; Tresserra-Rimbau, A.; Pintó, X.; Fitó, M.; Casas, R.; Martínez-González, M.Á.; Corella, D.; Salas-Salvadó, J.; Lapetra, J.; et al. Mediterranean diet and atherothrombosis biomarkers: A randomized controlled trial. Mol. Nutr. Food Res. 2020, 64, 2000350. [Google Scholar] [CrossRef]
- Ros, E.; Martínez-González, M.A.; Estruch, R.; Salas-Salvadó, J.; Fitó, M.; Martínez, J.A.; Corella, D. Mediterranean diet and cardiovascular health: Teachings of the PREDIMED study. Adv. Nutr. 2014, 5, 330S–336S. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Joshipura, K.J.; Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Rimm, E.B.; Speizer, F.E.; Colditz, G.; Ascherio, A.; Rosner, B.; Spiegelman, D.; et al. The effect of fruit and vegetable intake on risk for coronary heart disease. Ann. Intern. Med. 2001, 134, 1106–1114. [Google Scholar] [CrossRef]
- Fisher, M.; Levine, P.H.; Weiner, B.; Ockene, I.S.; Johnson, B.; Johnson, M.H.; Natale, A.M.; Vaudreuil, C.H.; Hoogasian, J. The effect of vegetarian diets on plasma lipid and platelet levels. Arch. Intern. Med. 1986, 146, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Rosell, M.S.; Lloyd-Wright, Z.; Appleby, P.N.; Sanders, T.A.B.; Allen, N.E.; Key, T.J. Long-chain n–3 polyunsaturated fatty acids in plasma in British meat-eating, vegetarian, and vegan men. Am. J. Clin. Nutr. 2005, 82, 327–334. [Google Scholar] [CrossRef]
- Bang, H.O.; Dyerberg, J. Lipid metabolism and ischemic heart disease in Greenland Eskimos. In Advances in Nutritional Research; Draper, H.H., Ed.; Springer: Boston, MA, USA, 1980; pp. 1–22. ISBN 978-1-4757-4450-7. [Google Scholar]
- Farsi, P.F.; Djazayery, A.; Eshraghian, M.R.; Koohdani, F.; Saboor-Yaraghi, A.A.; Derakhshanian, H.; Zarei, M.; Javanbakht, M.H.; Djalali, M. Effects of supplementation with omega-3 on insulin sensitivity and non-esterified free fatty acid (NEFA) in type 2 diabetic patients. Arq. Bras. Endocrinol. Metabol. 2014, 58, 335–340. [Google Scholar] [CrossRef]
- Selvaraj, S.; Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Juliano, R.A.; Jiao, L.; Tardif, J.; Ballantyne, C.M.; et al. Impact of icosapent ethyl on cardiovascular risk reduction in patients with heart failure in REDUCE-IT. J. Am. Heart Assoc. 2022, 11, e024999. [Google Scholar] [CrossRef]
- Yang, W.-S.; Chen, Y.-Y.; Chen, P.-C.; Hsu, H.-C.; Su, T.-C.; Lin, H.-J.; Chen, M.-F.; Lee, Y.-T.; Chien, K.-L. Association between plasma n-6 polyunsaturated fatty acids levels and the risk of cardiovascular disease in a community-based cohort study. Sci. Rep. 2019, 9, 19298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in plasma free fatty acids associated with type-2 diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [PubMed]
- Abell, T.L.; Minocha, A. Gastrointestinal complications of bariatric surgery: Diagnosis and therapy. Am. J. Med. Sci. 2006, 331, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Angrisani, L.; Santonicola, A.; Iovino, P.; Vitiello, A.; Zundel, N.; Buchwald, H.; Scopinaro, N. Bariatric surgery and endoluminal procedures: IFSO Worldwide Survey 2014. Obes. Surg. 2017, 27, 2279–2289. [Google Scholar] [CrossRef]
- Pucci, A.; Batterham, R.L. Mechanisms underlying the weight loss effects of RYGB and SG: Similar, yet different. J. Endocrinol. Investig. 2019, 42, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Pories, W.J. Bariatric surgery: Risks and rewards. J. Clin. Endocrinol. Metab. 2008, 93, s89–s96. [Google Scholar] [CrossRef]
- Buchwald, H.; Avidor, Y.; Braunwald, E.; Jensen, M.D.; Pories, W.; Fahrbach, K.; Schoelles, K. Bariatric surgery: A systematic review and meta-analysis. JAMA 2004, 292, 1724–1737. [Google Scholar] [CrossRef] [PubMed]
- Grenier-Larouche, T.; Carreau, A.-M.; Geloën, A.; Frisch, F.; Biertho, L.; Marceau, S.; Lebel, S.; Hould, F.-S.; Richard, D.; Tchernof, A.; et al. Fatty acid metabolic remodeling during type 2 diabetes remission after bariatric surgery. Diabetes 2017, 66, 2743–2755. [Google Scholar] [CrossRef] [PubMed]
- Nemati, R.; Lu, J.; Tura, A.; Smith, G.; Murphy, R. Acute Changes in Non-Esterified Fatty acids in patients with type 2 diabetes receiving bariatric surgery. Obes. Surg. 2017, 27, 649–656. [Google Scholar] [CrossRef]
- Sallé, A.; Demarsy, D.; Poirier, A.L.; Lelièvre, B.; Topart, P.; Guilloteau, G.; Bécouarn, G.; Rohmer, V. Zinc deficiency: A frequent and underestimated complication after bariatric surgery. Obes. Surg. 2010, 20, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Jasiñska, M.; Owczarek, J.; Orszulak-Michalak, D. Statins: A new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol. Rep. 2007, 59, 483–499. [Google Scholar]
- Campos, H.; Genest, J.J.; Blijlevens, E.; McNamara, J.R.; Jenner, J.L.; Ordovas, J.M.; Wilson, P.W.; Schaefer, E.J. Low density lipoprotein particle size and coronary artery disease. Arterioscler. Thromb. 1992, 12, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Statins and blood coagulation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 287–294. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot. Essent. Fat. Acids 2001, 65, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.I.; Hibbeln, J.R.; Mackey, R.H.; Muldoon, M.F. Statin treatment alters serum N-3 and n-6 fatty acids in hypercholesterolemic patients. Prostaglandins Leukot. Essent. Fat. Acids 2004, 71, 263–269. [Google Scholar] [CrossRef]
- van Raalte, D.H.; Li, M.; Pritchard, P.H.; Wasan, K.M. Peroxisome proliferator-activated receptor (PPAR) : A pharmacological target with a promising future. Pharm. Res. 2004, 21, 1531–1538. [Google Scholar] [CrossRef]
- Balakumar, P.; Mahadevan, N. Interplay between statins and PPARs in improving cardiovascular outcomes: A double-edged sword? Br. J. Pharmacol. 2012, 165, 373–379. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendía, L.E.; Pedone, C.; Ferretti, G.; Nachtigal, P.; Bo, S.; Derosa, G.; Maffioli, P.; Watts, G.F. Statin therapy and plasma free fatty acids: A systematic review and meta-analysis of controlled clinical trials: Statin therapy and free fatty acids. Br. J. Clin. Pharmacol. 2016, 81, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.Y.; Armstrong, P.C.J.; Dhanji, A.-R.A.; Tucker, A.T.; Paul-Clark, M.J.; Mitchell, J.A.; Warner, T.D. antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 706–711. [Google Scholar] [CrossRef]
- Blumenthal, R.S. Statins: Effective antiatherosclerotic therapy. Am. Heart J. 2000, 139, 577–583. [Google Scholar] [CrossRef]
- Grundy, S.M.; Vega, G.L.; Yuan, Z.; Battisti, W.P.; Brady, W.E.; Palmisano, J. Effectiveness and tolerability of simvastatin plus fenofibrate for combined hyperlipidemia (the SAFARI Trial). Am. J. Cardiol. 2005, 95, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Colli, S.; Eligini, S.; Lalli, M.; Camera, M.; Paoletti, R.; Tremoli, E. Vastatins inhibit tissue factor in cultured human macrophages. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 265–272. [Google Scholar] [CrossRef]
- Undas, A.; Kaczmarek, P.; Sladek, K.; Stepien, E.; Skucha, W.; Rzeszutko, M.; Gorkiewicz-Kot, I.; Tracz, W. Fibrin clot properties are altered in patients with chronic obstructive pulmonary disease. beneficial effects of simvastatin treatment. Thromb. Haemost. 2009, 102, 1176–1182. [Google Scholar] [PubMed]
- Undas, A.; Celinska-Löwenhoff, M.; Löwenhoff, T.; Szczeklik, A. Statins, fenofibrate, and quinapril increase clot permeability and enhance fibrinolysis in patients with coronary artery disease. J. Thromb. Haemost. 2006, 4, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Ghayour-Mobarhan, M.; Lamb, D.J.; Taylor, A.; Vaidya, N.; Livingstone, C.; Wang, T.; Ferns, G.A.A. effect of statin therapy on serum trace element status in dyslipidaemic subjects. J. Trace Elem. Med. Biol. 2005, 19, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Elisaf, M. Effects of fibrates on serum metabolic parameters. Curr. Med. Res. Opin. 2002, 18, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Vu-Dac, N.; Kosykh, V.A.; Saladin, R.; Fruchart, J.C.; Dallongeville, J.; Auwerx, J. Fibrates downregulate apolipoprotein c-iii expression independent of induction of peroxisomal acyl-coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J. Clin. Investig. 1995, 95, 705–712. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef]
- Martin, G.; Schoonjans, K.; Lefebvre, A.-M.; Staels, B.; Auwerx, J. Coordinate regulation of the expression of the fatty acid transport protein and Acyl-CoA synthetase genes by PPARα and PPARγ activators. J. Biol. Chem. 1997, 272, 28210–28217. [Google Scholar] [CrossRef]
- Heller, F.; Harvengt, C. Effects of clofibrate, bezafibrate, fenofibrate and probucol on plasma lipolytic enzymes in normolipaemic subjects. Eur. J. Clin. Pharmacol. 1983, 25, 57–63. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.-A. Is there a role for fibrates in the management of dyslipidemia in the metabolic syndrome? Arterioscler. Thromb. Vasc. Biol. 2008, 28, 39–46. [Google Scholar] [CrossRef]
- Jun, M.; Foote, C.; Lv, J.; Neal, B.; Patel, A.; Nicholls, S.J.; Grobbee, D.E.; Cass, A.; Chalmers, J.; Perkovic, V. Effects of fibrates on cardiovascular outcomes: A systematic review and meta-analysis. Lancet 2010, 375, 1875–1884. [Google Scholar] [CrossRef]
- Malur, P.; Menezes, A.; DiNicolantonio, J.J.; O’Keefe, J.H.; Lavie, C.J. The microvascular and macrovascular benefits of fibrates in diabetes and the metabolic syndrome: A review. Mo. Med. 2017, 114, 464–471. [Google Scholar] [PubMed]
- Creager, M.A.; Lüscher, T.F.; prepared with the assistance of Cosentino, F. and Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., III; Klein, R.; for the American Diabetes Association. Retinopathy in Diabetes. Diabetes Care 2004, 27, s84–s87. [Google Scholar] [CrossRef] [PubMed]
- Hadjivasilis, A.; Kouis, P.; Kousios, A.; Panayiotou, A. the effect of fibrates on kidney function and chronic kidney disease progression: A systematic review and meta-analysis of randomised studies. J. Clin. Med. 2022, 11, 768. [Google Scholar] [CrossRef]
- Klen, J.; Dolžan, V. Glucagon-like Peptide-1 Receptor agonists in the management of type 2 diabetes mellitus and obesity: The impact of pharmacological properties and genetic factors. Int. J. Mol. Sci. 2022, 23, 3451. [Google Scholar] [CrossRef]
- McLean, B.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Anholm, C.; Kumarathurai, P.; Samkani, A.; Pedersen, L.R.; Boston, R.C.; Nielsen, O.W.; Kristiansen, O.P.; Fenger, M.; Madsbad, S.; Sajadieh, A.; et al. Effect of liraglutide on estimates of lipolysis and lipid oxidation in obese patients with stable coronary artery disease and newly diagnosed type 2 diabetes: A randomized trial. Diabetes Obes. Metab. 2019, 21, 2012–2016. [Google Scholar] [CrossRef]
- Jendle, J.; Hyötyläinen, T.; Orešič, M.; Nyström, T. Pharmacometabolomic profiles in type 2 diabetic subjects treated with liraglutide or glimepiride. Cardiovasc. Diabetol. 2021, 20, 237. [Google Scholar] [CrossRef]
- Koska, J.; Lopez, L.; D’Souza, K.; Osredkar, T.; Deer, J.; Kurtz, J.; Salbe, A.D.; Harman, S.M.; Reaven, P.D. Effect of Liraglutide on dietary lipid-induced insulin resistance in Humans. Diabetes Obes. Metab. 2018, 20, 69–76. [Google Scholar] [CrossRef]
- Smith, S.; Witkowski, A.; Joshi, A.K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003, 42, 289–317. [Google Scholar] [CrossRef]
- Cha, S.H.; Hu, Z.; Chohnan, S.; Lane, M.D. Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc. Natl. Acad. Sci. USA 2005, 102, 14557–14562. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty acid synthase and cancer: New application of an old pathway. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- Morak, M.; Schmidinger, H.; Riesenhuber, G.; Rechberger, G.N.; Kollroser, M.; Haemmerle, G.; Zechner, R.; Kronenberg, F.; Hermetter, A. Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) deficiencies affect expression of lipolytic activities in mouse adipose tissues. Mol. Cell. Proteom. 2012, 11, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Langin, D. Hormone-sensitive lipase deficiency in humans. Cell Metab. 2014, 20, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Piché, M.-E.; Parry, S.A.; Karpe, F.; Hodson, L. Chylomicron-derived fatty acid spillover in adipose tissue: A signature of metabolic health? J. Clin. Endocrinol. Metab. 2018, 103, 25–34. [Google Scholar] [CrossRef]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2010, 60, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerciolini, R. Mode of action of Orlistat. Int. J. Obes. Relat. Metab. Disord. 1997, 21 (Suppl. S3), S12–S23. [Google Scholar] [PubMed]
- Heck, A.M.; Yanovski, J.A.; Calis, K.A. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy 2000, 20, 270–279. [Google Scholar] [CrossRef]
- Malong, C.; Delgado, J.; Cunanan, E. Efficacy of orlistat vs placebo in the improvement of lipid profile among overweight and obese patients: A meta-analysis. Endocr. Abst. 2012, 29, P1185. [Google Scholar]
- Di Somma, C.; Rivellese, A.; Pizza, G.; Patti, L.; De Rosa, A.; Cipriano, P.; Nedi, V.; Rossi, A.; Lombardi, G.; Colao, A.; et al. Effects of short-term treatment with Orlistat on growth hormone/insulin-like growth factor-I axis in obese post-menopausal women. J. Endocrinol. Investig. 2011, 34, 90–96. [Google Scholar] [CrossRef]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Hesler, C.B.; Swenson, T.L.; Tall, A.R. Purification and characterization of a human plasma cholesteryl ester transfer protein. J. Biol. Chem. 1987, 262, 2275–2282. [Google Scholar] [CrossRef]
- Taheri, H.; Filion, K.B.; Windle, S.B.; Reynier, P.; Eisenberg, M.J. Cholesteryl ester transfer protein inhibitors and cardiovascular outcomes: A systematic review and meta-analysis of randomized controlled trials. Cardiology 2020, 145, 236–250. [Google Scholar] [CrossRef]
- Briand, F.; Prunet-Marcassus, B.; Thieblemont, Q.; Costard, C.; Muzotte, E.; Sordello, S.; Sulpice, T. Raising HDL with CETP inhibitor Torcetrapib improves glucose homeostasis in dyslipidemic and insulin resistant hamsters. Atherosclerosis 2014, 233, 359–362. [Google Scholar] [CrossRef]
- Villarruz-Sulit, M.V.; Forster, R.; Dans, A.L.; Tan, F.N.; Sulit, D.V. Chelation therapy for atherosclerotic cardiovascular disease. Cochrane Database Syst. Rev. 2020, 5, CD002785. [Google Scholar]
- Flora, S.J.S.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Lindblad, L.; Lewis, E.F.; Drisko, J.; Lee, K.L.; et al. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: The TACT randomized trial. JAMA 2013, 309, 1241. [Google Scholar] [CrossRef] [PubMed]
- Escolar, E.; Ujueta, F.; Kim, H.; Mark, D.B.; Boineau, R.; Nahin, R.L.; Goertz, C.; Lee, K.L.; Anstrom, K.J.; Lamas, G.A. Possible differential benefits of edetate disodium in post-myocardial infarction patients with diabetes treated with different hypoglycemic strategies in the trial to assess chelation therapy (TACT). J. Diabetes Its Complicat. 2020, 34, 107616. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, J.A.C.; Daniel, C. Harris: Quantitative chemical analysis, 9th Ed. Anal. Bioanal. Chem. 2015, 407, 8943–8944. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Z.; Li, Y.V. Metal Ion chelation enhances tissue plasminogen activator (TPA)-induced thrombolysis: An in vitro and in vivo study. J. Thromb. Thrombolysis 2022, 53, 291–301. [Google Scholar] [CrossRef]
- Lu, J.; Stewart, A.J.; Sleep, D.; Sadler, P.J.; Pinheiro, T.J.T.; Blindauer, C.A. A molecular mechanism for modulating plasma Zn speciation by fatty acids. J. Am. Chem. Soc. 2012, 134, 1454–1457. [Google Scholar] [CrossRef]
- Al-Harthi, S.; Chandra, K.; Jaremko, Ł. Lipoic Acid restores binding of Zinc ions to human serum albumin. Front. Chem. 2022, 10, 942585. [Google Scholar] [CrossRef] [PubMed]
- Leboffe, L.; di Masi, A.; Trezza, V.; Polticelli, F.; Ascenzi, P. Human serum albumin: A modulator of cannabinoid drugs. IUBMB Life 2017, 69, 834–840. [Google Scholar] [CrossRef]
- Peters, T., Jr. All about albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: New York, NY, USA, 1995; ISBN 978-0-12-552110-9. [Google Scholar]
- Richardson, V.R.; Schroeder, V.; Grant, P.J.; Standeven, K.F.; Carter, A.M. Complement C3 is a substrate for activated factor xiii that is cross-linked to fibrin during clot formation. Br. J. Haematol. 2013, 160, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.; Alzahrani, S.H.; Price, J.F.; Strachan, M.W.; Oxley, N.; King, R.; Gamlen, T.; Schroeder, V.; Baxter, P.D.; Ajjan, R.A. Hypofibrinolysis in type 2 diabetes: The role of the inflammatory pathway and complement C3. Diabetologia 2014, 57, 1737–1741. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Tiede, C.; Simmons, K.; Fishwick, C.; Tomlinson, D.; Ajjan, R. Inhibition of complement C3 and fibrinogen interaction: A potential novel therapeutic target to reduce cardiovascular disease in diabetes. Lancet 2015, 385, S57. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regan-Smith, S.; Fritzen, R.; Hierons, S.J.; Ajjan, R.A.; Blindauer, C.A.; Stewart, A.J. Strategies for Therapeutic Amelioration of Aberrant Plasma Zn2+ Handling in Thrombotic Disease: Targeting Fatty Acid/Serum Albumin-Mediated Effects. Int. J. Mol. Sci. 2022, 23, 10302. https://doi.org/10.3390/ijms231810302
Regan-Smith S, Fritzen R, Hierons SJ, Ajjan RA, Blindauer CA, Stewart AJ. Strategies for Therapeutic Amelioration of Aberrant Plasma Zn2+ Handling in Thrombotic Disease: Targeting Fatty Acid/Serum Albumin-Mediated Effects. International Journal of Molecular Sciences. 2022; 23(18):10302. https://doi.org/10.3390/ijms231810302
Chicago/Turabian StyleRegan-Smith, Spencer, Remi Fritzen, Stephen J. Hierons, Ramzi A. Ajjan, Claudia A. Blindauer, and Alan J. Stewart. 2022. "Strategies for Therapeutic Amelioration of Aberrant Plasma Zn2+ Handling in Thrombotic Disease: Targeting Fatty Acid/Serum Albumin-Mediated Effects" International Journal of Molecular Sciences 23, no. 18: 10302. https://doi.org/10.3390/ijms231810302