
Renal Transcriptome and Metabolome in Mice with Principal Cell-Specific Ablation of the Tsc1 Gene: Derangements in Pathways Associated with Cell Metabolism, Growth and Acid Secretion

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Principal Cell-Specific Ablation of the Tsc1 Gene
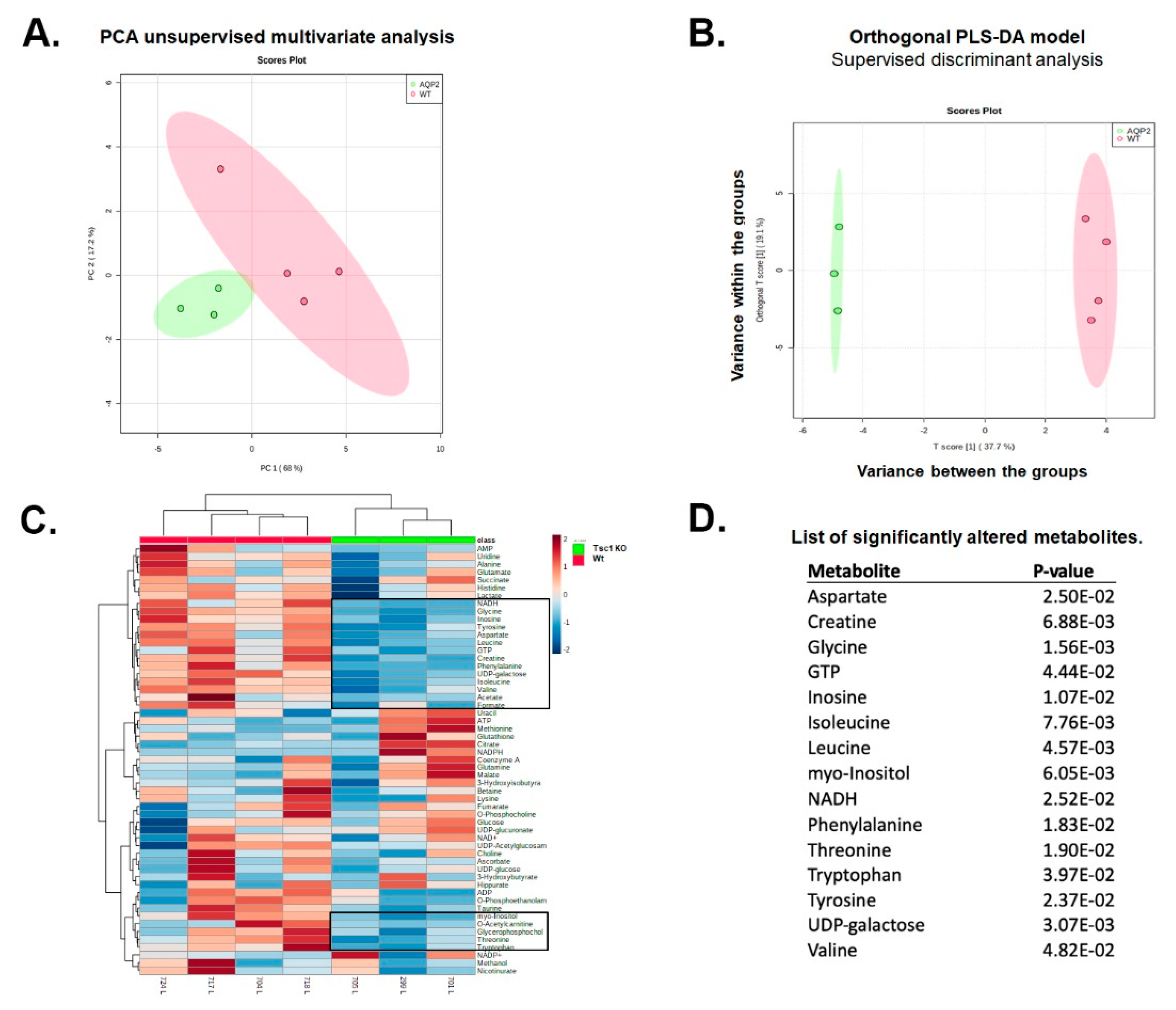
2.2. 1H NMR Analysis of Extracts from Kidneys of Wt and Tsc1 KO Mice
2.3. Analysis of 1H NMR Spectroscopic Data
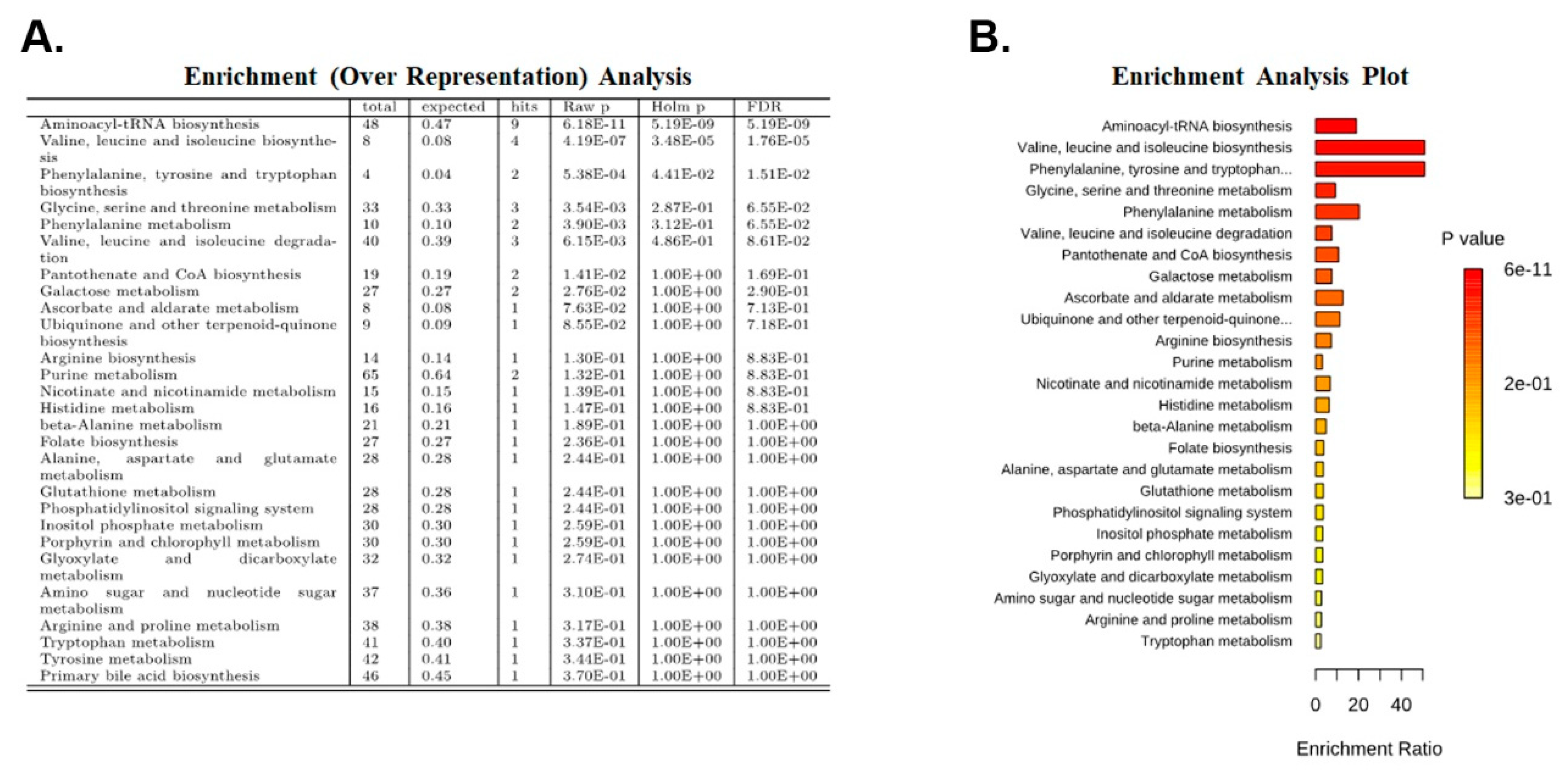
2.4. Enrichment Analysis
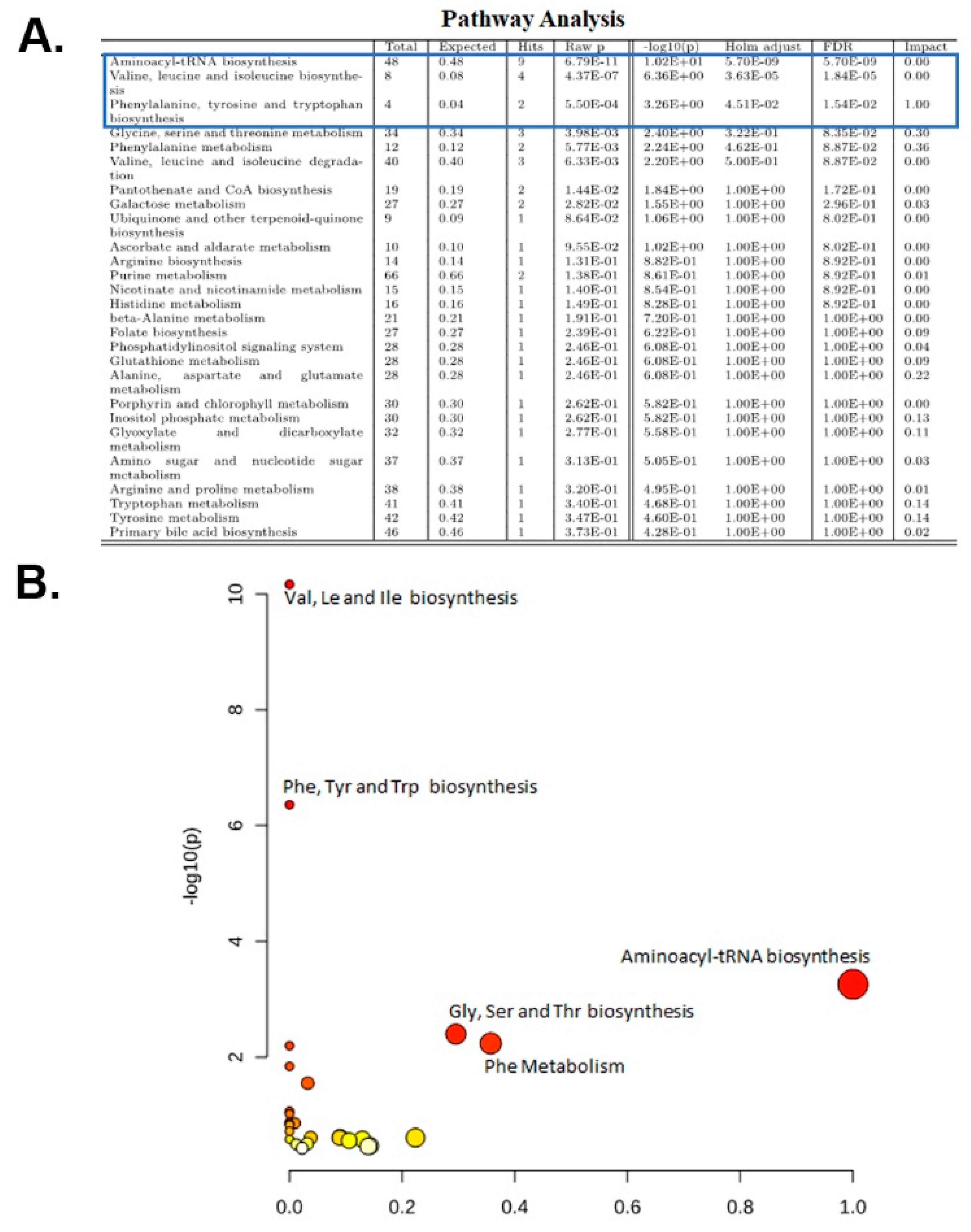
2.5. Pathway Analysis
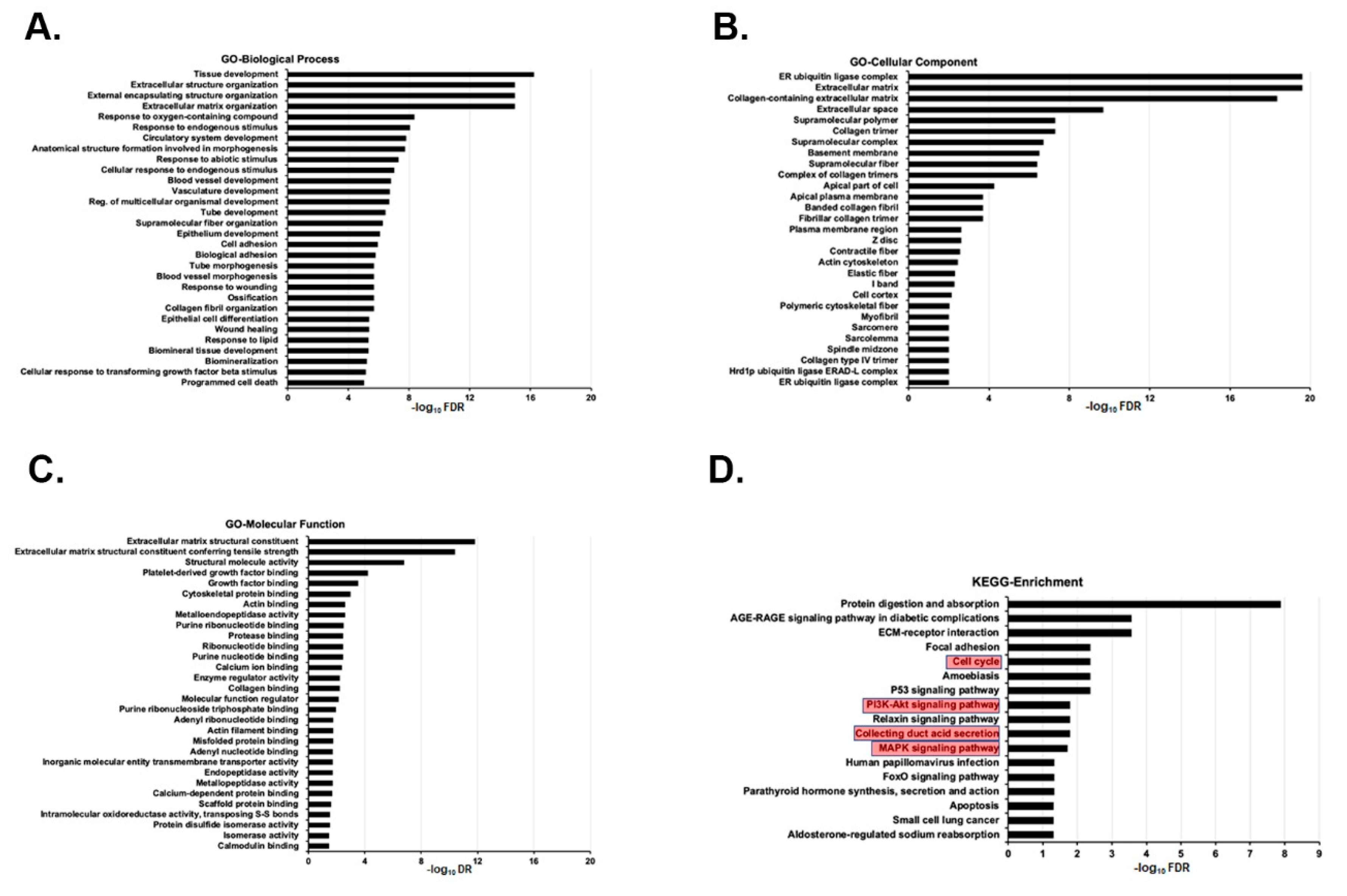
2.6. Kidneys of Tsc1 KO Mice Have a Distinct Transcriptome Fingerprint Compared to Those of Wt Mice
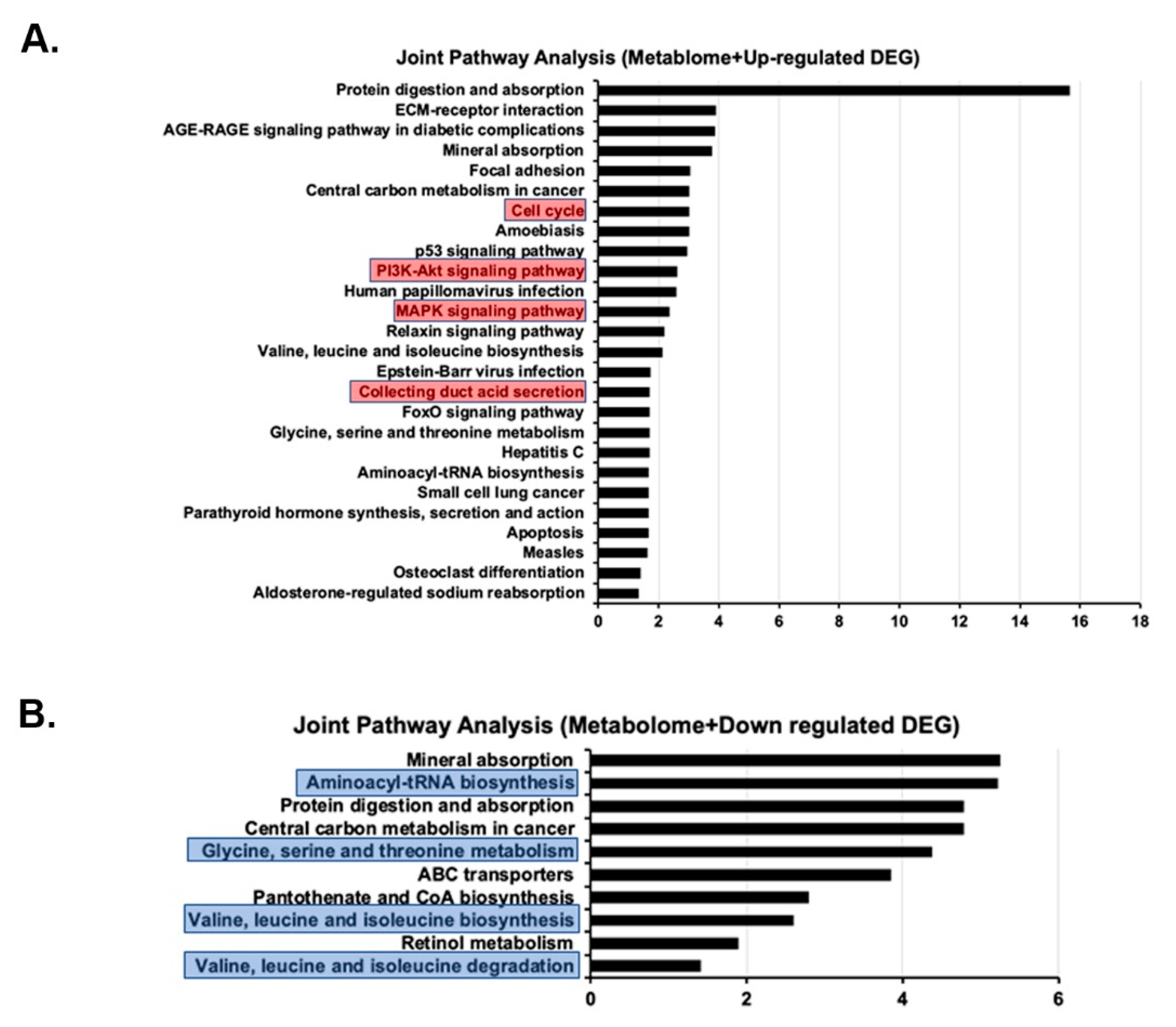
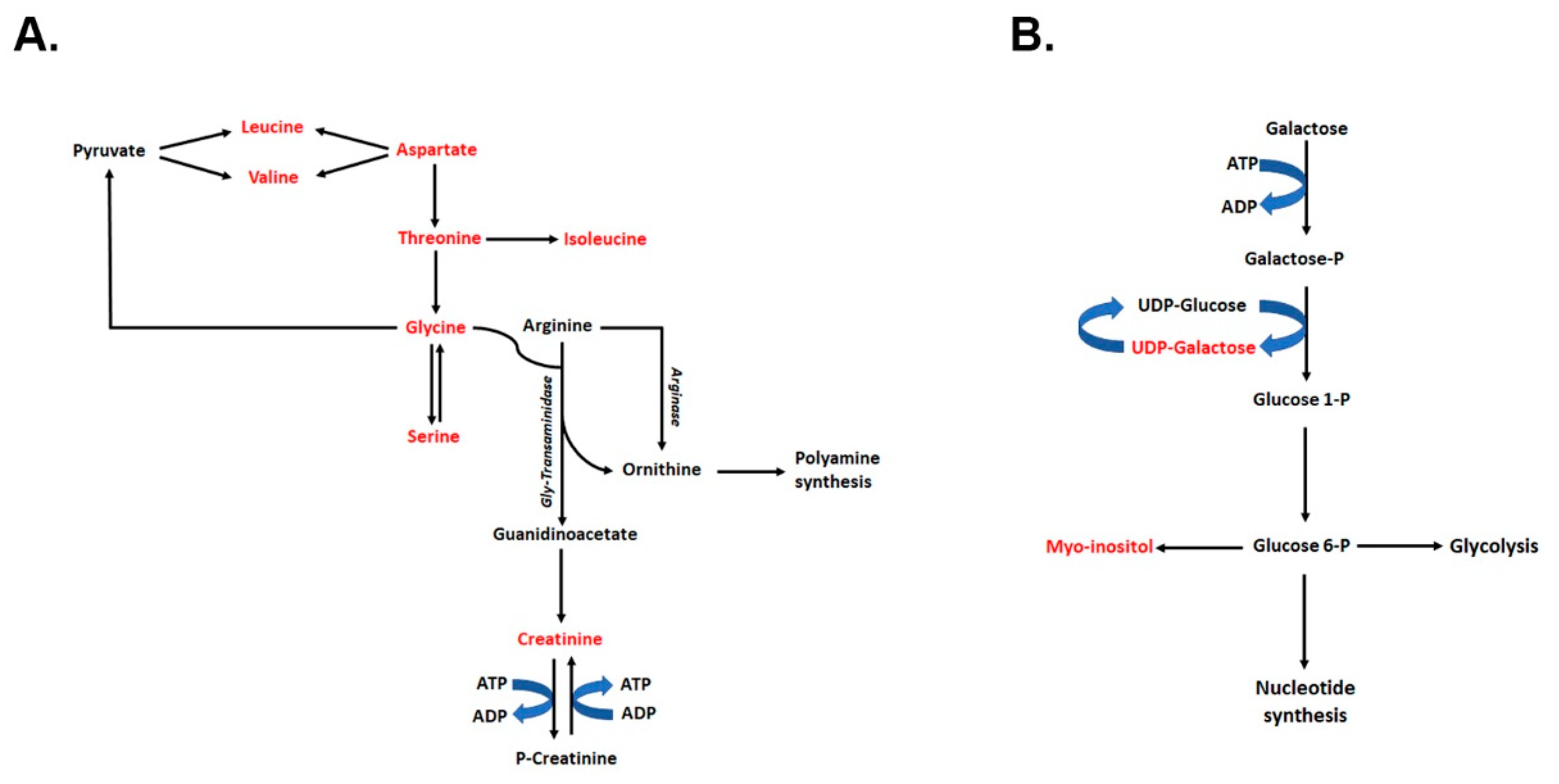
2.7. Joint Pathway Analysis of Metabolome and Transcriptome Results
2.8. Polyamines Levels and Their Synthesis Are Not Affected in the Kidneys of Tsc1 KO Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Tissue Collection
4.4. Extraction of Metabolites
4.5. RNA Extraction
4.6. NMR Sample Preparation and Spectra Acquisition
4.7. RNA-seq Analysis
4.8. Data Processing and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.; Halpern, E.F.; Thiele, E.A. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef]
- Lam, H.C.; Siroky, B.J.; Henske, E.P. Renal disease in tuberous sclerosis complex: Pathogenesis and therapy. Nat. Rev. Nephrol. 2018, 14, 704–716. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Long, X.; Ortiz-Vega, S.; Lin, Y.; Avruch, J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 2005, 280, 23433–23436. [Google Scholar] [CrossRef] [Green Version]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.J. Human Protein and Amino Acid Requirements. J. Parenter. Enter. Nutr. 2016, 40, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, Y.; Nakamura, N.; Ikeda, N.; Sugiyama, R.; Ishii, C.; Maki, M.; Shibata, H.; Takahara, T. Amino Acid-Mediated Intracellular Ca(2+) Rise Modulates mTORC1 by Regulating the TSC2-Rheb Axis through Ca(2+)/Calmodulin. Int. J. Mol. Sci. 2021, 22, 6897. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Ting, C.Y.; Bettedi, L.; Kim, K.; Ghaniam, E.; Lilly, M.A. The Rag GTPase Regulates the Dynamic Behavior of TSC Downstream of Both Amino Acid and Growth Factor Restriction. Dev. Cell 2020, 55, 272–288.e5. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef]
- McKenna, J., 3rd; Kapfhamer, D.; Kinchen, J.M.; Wasek, B.; Dunworth, M.; Murray-Stewart, T.; Bottiglieri, T.; Casero, R.A., Jr.; Gambello, M.J. Metabolomic studies identify changes in transmethylation and polyamine metabolism in a brain-specific mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2018, 27, 2113–2124. [Google Scholar] [CrossRef]
- Tang, Y.; El-Chemaly, S.; Taveira-Dasilva, A.; Goldberg, H.J.; Bagwe, S.; Rosas, I.O.; Moss, J.; Priolo, C.; Henske, E.P. Alterations in Polyamine Metabolism in Patients With Lymphangioleiomyomatosis and Tuberous Sclerosis Complex 2-Deficient Cells. Chest 2019, 156, 1137–1148. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [Green Version]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Lux, A.; Calder, N.; Laugharne, M.; Osborne, J.; O’Callaghan, F. Causes of mortality in individuals with tuberous sclerosis complex. Dev. Med. Child Neurol. 2017, 59, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.W.; Gomez, M.R.; Lie, J.T.; Crowson, C.S. Causes of death in patients with tuberous sclerosis. Mayo Clin. Proc. 1991, 66, 792–796. [Google Scholar] [CrossRef]
- Barone, S.; Zahedi, K.; Brooks, M.; Henske, E.P.; Yang, Y.; Zhang, E.; Bissler, J.J.; Yu, J.J.; Soleimani, M. Kidney intercalated cells and the transcription factor FOXi1 drive cystogenesis in tuberous sclerosis complex. Proc Natl. Acad. Sci. USA 2021, 118, e2020190118. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Zadjali, F.; Bridges, D.; Astrinidis, A.; Barone, S.; Yao, Y.; Redd, J.R.; Siroky, B.J.; Wang, Y.; Finley, J.T.; et al. Tuberous sclerosis complex exhibits a new renal cystogenic mechanism. Physiol. Rep. 2019, 7, e13983. [Google Scholar] [CrossRef]
- An, J.N.; Hyeon, J.S.; Jung, Y.; Choi, Y.W.; Kim, J.H.; Yang, S.H.; Oh, S.; Kwon, S.; Lee, S.H.; Cho, J.H.; et al. Urinary myo-inositol is associated with the clinical outcome in focal segmental glomerulosclerosis. Sci. Rep. 2019, 9, 14707. [Google Scholar] [CrossRef]
- Chang, H.H.; Chao, H.N.; Walker, C.S.; Choong, S.Y.; Phillips, A.; Loomes, K.M. Renal depletion of myo-inositol is associated with its increased degradation in animal models of metabolic disease. Am. J. Physiol. Renal Physiol. 2015, 309, F755–F763. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A.; Dinicola, S.; Harrath, A.H.; Alwasel, S.H.; Unfer, V.; Bevilacqua, A. Does myo-inositol effect on PCOS follicles involve cytoskeleton regulation? Med. Hypotheses 2016, 91, 1–5. [Google Scholar] [CrossRef]
- Monastra, G.; Unfer, V.; Harrath, A.H.; Bizzarri, M. Combining treatment with myo-inositol and D-chiro-inositol (40:1) is effective in restoring ovary function and metabolic balance in PCOS patients. Gynecol. Endocrinol. 2017, 33, 1–9. [Google Scholar] [CrossRef]
- Napolioni, V.; Curatolo, P. Genetics and molecular biology of tuberous sclerosis complex. Curr. Genom. 2008, 9, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Sofia, A.; Saffioti, S.; Bonanni, A.; Mannucci, I.; Verzola, D. Amino acid and protein metabolism in the human kidney and in patients with chronic kidney disease. Clin. Nutr. 2010, 29, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Picciotto, D.; Saio, M.; Aimasso, F.; Bruzzone, F.; Sukkar, S.G.; Massarino, F.; Esposito, P.; Viazzi, F.; Garibotto, G. Low Protein Diets and Plant-Based Low Protein Diets: Do They Meet Protein Requirements of Patients with Chronic Kidney Disease? Nutrients 2020, 13, 83. [Google Scholar] [CrossRef]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef]
- Broussard, A.; Florwick, A.; Desbiens, C.; Nischan, N.; Robertson, C.; Guan, Z.; Kohler, J.J.; Wells, L.; Boyce, M. Human UDP-galactose 4’-epimerase (GALE) is required for cell-surface glycome structure and function. J. Biol. Chem. 2020, 295, 1225–1239. [Google Scholar] [CrossRef]
- Broussard, A.C.; Boyce, M. Life is sweet: The cell biology of glycoconjugates. Mol. Biol. Cell 2019, 30, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Kabra, P.M.; Lee, H.K.; Lubich, W.P.; Marton, L.J. Solid-phase extraction and determination of dansyl derivatives of unconjugated and acetylated polyamines by reversed-phase liquid chromatography: Improved separation systems for polyamines in cerebrospinal fluid, urine and tissue. J. Chromatogr. 1986, 380, 19–32. [Google Scholar] [CrossRef]
- Porter, C.W.; Ganis, B.; Libby, P.R.; Bergeron, R.J. Correlations between polyamine analogue-induced increases in spermidine/spermine N1-acetyltransferase activity, polyamine pool depletion, and growth inhibition in human melanoma cell lines. Cancer Res. 1991, 51, 3715–3720. [Google Scholar]
- Seely, J.E.; Pegg, A.E. Changes in mouse kidney ornithine decarboxylase activity are brought about by changes in the amount of enzyme protein as measured by radioimmunoassay. J. Biol. Chem. 1983, 258, 2496–2500. [Google Scholar] [CrossRef]
- Seely, J.E.; Pegg, A.E. Ornithine decarboxylase (mouse kidney). Methods Enzymol. 1983, 94, 158–161. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahedi, K.; Barone, S.; Brooks, M.; Murray Stewart, T.; Casero, R.A., Jr.; Soleimani, M. Renal Transcriptome and Metabolome in Mice with Principal Cell-Specific Ablation of the Tsc1 Gene: Derangements in Pathways Associated with Cell Metabolism, Growth and Acid Secretion. Int. J. Mol. Sci. 2022, 23, 10601. https://doi.org/10.3390/ijms231810601
Zahedi K, Barone S, Brooks M, Murray Stewart T, Casero RA Jr., Soleimani M. Renal Transcriptome and Metabolome in Mice with Principal Cell-Specific Ablation of the Tsc1 Gene: Derangements in Pathways Associated with Cell Metabolism, Growth and Acid Secretion. International Journal of Molecular Sciences. 2022; 23(18):10601. https://doi.org/10.3390/ijms231810601
Chicago/Turabian StyleZahedi, Kamyar, Sharon Barone, Marybeth Brooks, Tracy Murray Stewart, Robert A. Casero, Jr., and Manoocher Soleimani. 2022. "Renal Transcriptome and Metabolome in Mice with Principal Cell-Specific Ablation of the Tsc1 Gene: Derangements in Pathways Associated with Cell Metabolism, Growth and Acid Secretion" International Journal of Molecular Sciences 23, no. 18: 10601. https://doi.org/10.3390/ijms231810601
APA StyleZahedi, K., Barone, S., Brooks, M., Murray Stewart, T., Casero, R. A., Jr., & Soleimani, M. (2022). Renal Transcriptome and Metabolome in Mice with Principal Cell-Specific Ablation of the Tsc1 Gene: Derangements in Pathways Associated with Cell Metabolism, Growth and Acid Secretion. International Journal of Molecular Sciences, 23(18), 10601. https://doi.org/10.3390/ijms231810601