
GB83, an Agonist of PAR2 with a Unique Mechanism of Action Distinct from Trypsin and PAR2-AP

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
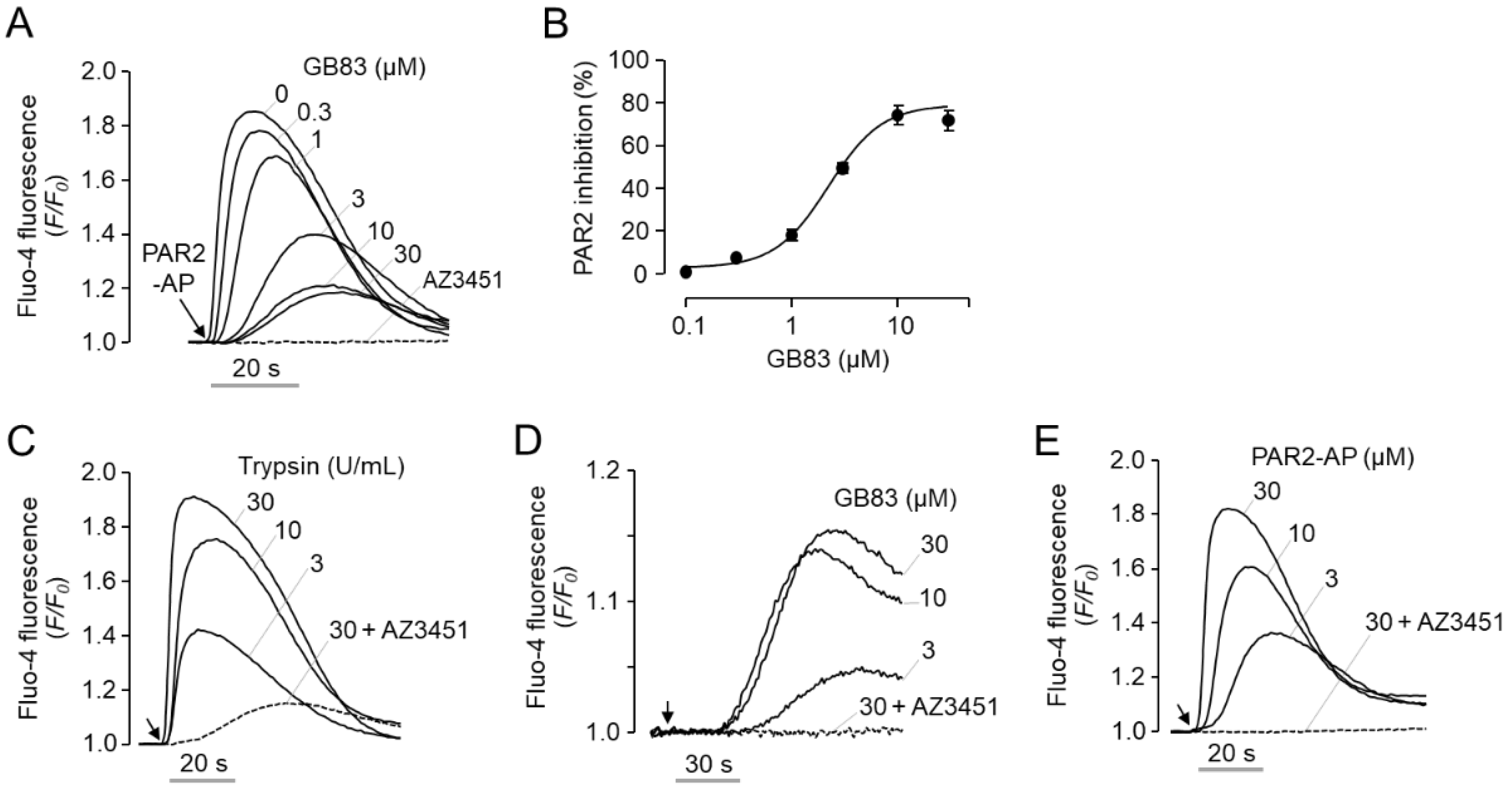
2.1. GB83 Is a Bona Fide PAR2 Agonist and Induces Elevation of Intracellular Calcium through Activation of PAR2
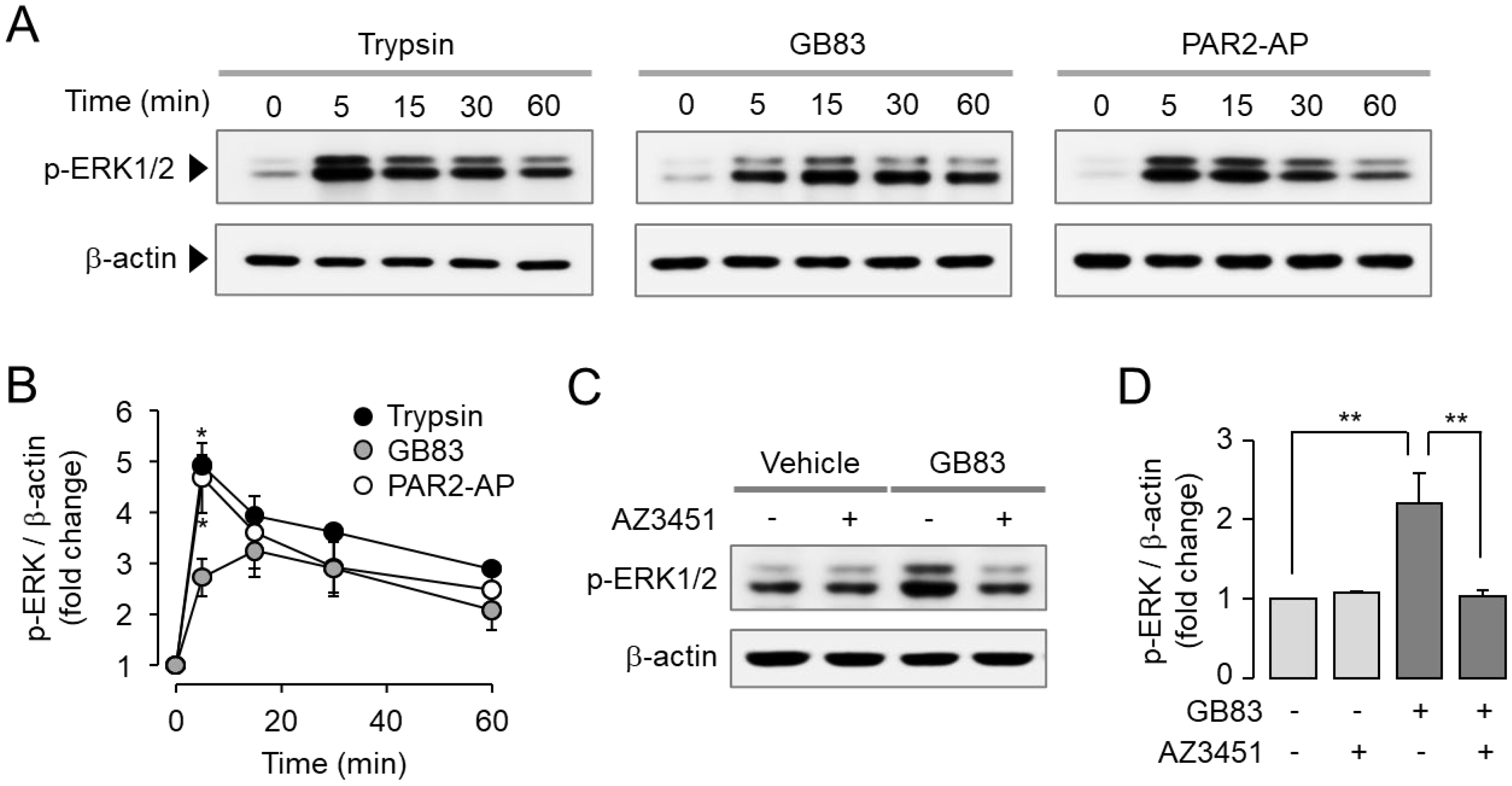
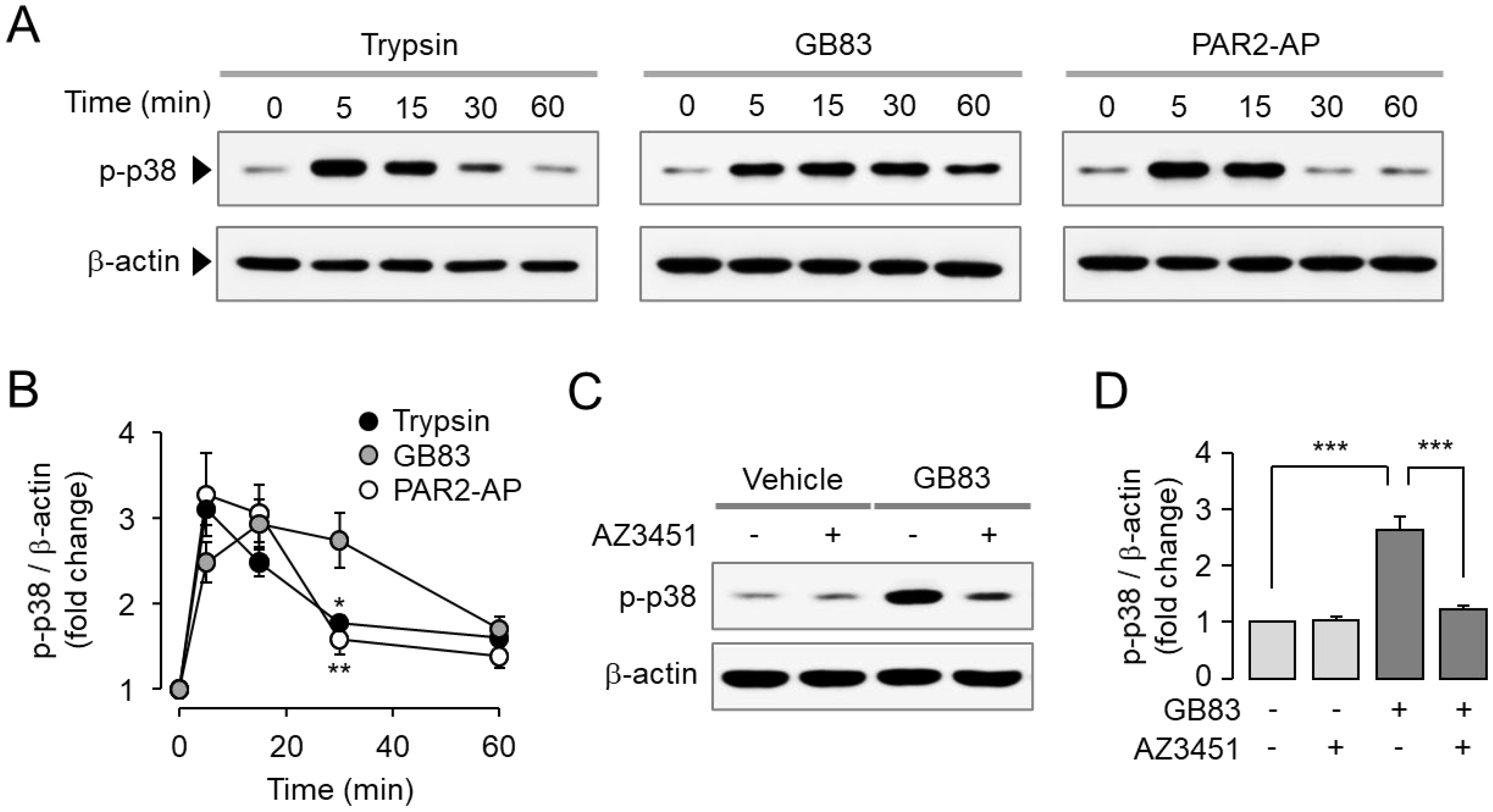
2.2. GB83 Induces PAR2-Mediated Phosphorylation of ERK1/2 and p38
2.3. GB83 Induces Sustained Endocytosis of PAR2
2.4. GB83 Induces Sustained Colocalization of PAR2 and β-Arrestins
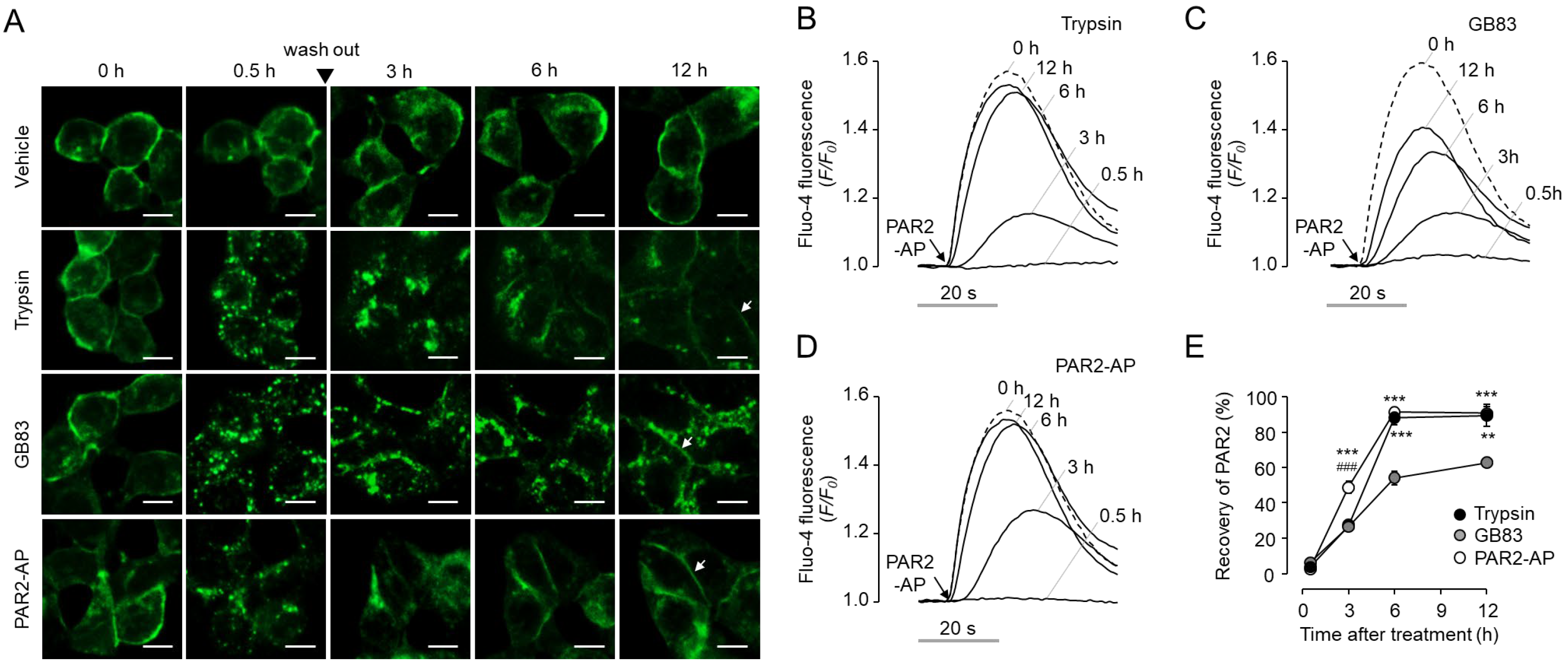
2.5. GB83 Induces Prolonged PAR2 Desensitization and Slow Recovery of Cell Surface PAR2
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Materials and Reagents
4.3. Molecular Cloning of Plasmid Constructs
4.4. Generation of Stable Cell Line
4.5. Intracellular Calcium Measurement
4.6. Immunoblotting
4.7. Live Cell Imaging
4.8. Quantification of Colocalization
4.9. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | Activating peptide |
| GPCR | G-protein-coupled receptor |
| PAR | Protease-activated receptor |
| PCC | Pearson’s correlation coefficient |
References
- Schmidlin, F.; Amadesi, S.; Vidil, R.; Trevisani, M.; Martinet, N.; Caughey, G.; Tognetto, M.; Cavallesco, G.; Mapp, C.; Geppetti, P.; et al. Expression and function of proteinase-activated receptor 2 in human bronchial smooth muscle. Am. J. Respir. Crit. Care Med. 2001, 164, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Rattenholl, A.; Steinhoff, M. Proteinase-activated receptor-2 in the skin: Receptor expression, activation and function during health and disease. Drug News Perspect. 2008, 21, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Vergnolle, N. Clinical relevance of proteinase activated receptors (pars) in the gut. Gut 2005, 54, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, D.; Schuepbach, R. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 1–24. [Google Scholar] [CrossRef]
- Frungieri, M.; Weidinger, S.; Meineke, V.; Köhn, F.; Mayerhofer, A. Proliferative action of mast-cell tryptase is mediated by PAR2, COX2, prostaglandins, and PPARgamma: Possible relevance to human fibrotic disorders. Proc. Natl. Acad. Sci. USA 2002, 99, 15072–15077. [Google Scholar] [CrossRef]
- Rothmeier, A.; Liu, E.; Chakrabarty, S.; Disse, J.; Mueller, B.; Østergaard, H.; Ruf, W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 2018, 131, 674–685. [Google Scholar] [CrossRef]
- Johnson, J.J.; Miller, D.L.; Jiang, R.; Liu, Y.; Shi, Z.; Tarwater, L.; Williams, R.; Balsara, R.; Sauter, E.R.; Stack, M.S. Protease-activated Receptor-2 (PAR-2)-mediated Nf-κB Activation Suppresses Inflammation-associated Tumor Suppressor MicroRNAs in Oral Squamous Cell Carcinoma. J. Biol. Chem. 2016, 291, 6936–6945. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.-S.; Rhee, M.H.; Sung, G.-H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 2014, 352371. [Google Scholar] [CrossRef]
- Fyfe, M.; Bergström, M.; Aspengren, S.; Peterson, A. PAR-2 activation in intestinal epithelial cells potentiates interleukin-1beta-induced chemokine secretion via MAP kinase signaling pathways. Cytokine 2005, 31, 358–367. [Google Scholar] [CrossRef]
- Frateschi, S.; Camerer, E.; Crisante, G.; Rieser, S.; Membrez, M.; Charles, R.; Beermann, F.; Stehle, J.; Breiden, B.; Sandhoff, K.; et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat. Commun. 2011, 2, 161. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Ni, B.; Xi, Y.; Chu, X.; Zhang, R.; You, H. Protease-activated receptor 2 (PAR-2) antagonist AZ3451 as a novel therapeutic agent for osteoarthritis. Aging 2019, 11, 12532–12545. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.; Jeong, S.; Hong, S. Protease-Activated Receptors 2-Antagonist Suppresses Asthma by Inhibiting Reactive Oxygen Species-Thymic Stromal Lymphopoietin Inflammation and Epithelial Tight Junction Degradation. Allergy Asthma Immunol. Res. 2019, 11, 560–571. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, K.; McGrath, S.; Huesa, C.; Dunning, L.; Litherland, G.; Crilly, A.; Hultin, L.; Ferrell, W.; Lockhart, J.; Goodyear, C. Rheumatic Disease: Protease-Activated Receptor-2 in Synovial Joint Pathobiology. Front. Endocrinol. 2018, 9, 00257. [Google Scholar] [CrossRef] [PubMed]
- Sébert, M.; Sola-Tapias, N.; Mas, E.; Barreau, F.; Ferrand, A. Protease-Activated Receptors in the Intestine: Focus on Inflammation and Cancer. Front. Endocrinol. 2019, 10, 717. [Google Scholar] [CrossRef]
- Subramaniam, S.; Ruf, W.; Bosmann, M. Advocacy of targeting protease-activated receptors in severe coronavirus disease 2019. Br. J. Pharmacol. 2021, 179, 2086–2099. [Google Scholar] [CrossRef]
- Rajagopal, S.; Shenoy, S. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef]
- Tian, X.; Kang, D.; Benovic, J. β-arrestins and G protein-coupled receptor trafficking. Handb. Exp. Pharmacol. 2014, 219, 173–186. [Google Scholar] [CrossRef]
- Déry, O.; Thoma, M.; Wong, H.; Grady, E.; Bunnett, N. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-Arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 1999, 274, 18524–18535. [Google Scholar] [CrossRef]
- Goh, F.; Ng, P.; Nilsson, M.; Kanke, T.; Plevin, R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br. J. Pharmacol. 2009, 158, 1695–1704. [Google Scholar] [CrossRef]
- Boitano, S.; Hoffman, J.; Flynn, A.; Asiedu, M.; Tillu, D.; Zhang, Z.; Sherwood, C.; Rivas, C.; DeFea, K.; Vagner, J.; et al. The novel PAR2 ligand C391 blocks multiple PAR2 signalling pathways in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Suen, J.; Cotterell, A.; Lohman, R.; Lim, J.; Han, A.; Yau, M.; Liu, L.; Cooper, M.; Vesey, D.; Fairlie, D. Pathway-selective antagonism of proteinase activated receptor 2. Br. J. Pharmacol. 2014, 171, 4112–4124. [Google Scholar] [CrossRef]
- Barry, G.; Suen, J.; Le, G.; Cotterell, A.; Reid, R.; Fairlie, D. Novel agonists and antagonists for human protease activated receptor 2. J. Med. Chem. 2010, 53, 7428–7440. [Google Scholar] [CrossRef]
- Pierre, O.; Fouchard, M.; Buscaglia, P.; Goux, N.L.; Leschiera, R.; Mignen, O.; Fluhr, J.; Misery, L.; Garrec, R.L. Calcium Increase and Substance P Release Induced by the Neurotoxin Brevetoxin-1 in Sensory Neurons: Involvement of PAR2 Activation through Both Cathepsin S and Canonical Signaling. Cells 2020, 9, 2704. [Google Scholar] [CrossRef]
- Muley, M.; Reid, A.; Botz, B.; Bölcskei, K.; Helyes, Z.; McDougall, J. Neutrophil elastase induces inflammation and pain in mouse knee joints via activation of proteinase-activated receptor-2. Br. J. Pharmacol. 2016, 173, 766–777. [Google Scholar] [CrossRef]
- Indrakusuma, I.; Romacho, T.; Eckel, J. Protease-Activated Receptor 2 Promotes Pro-Atherogenic Effects through Transactivation of the VEGF Receptor 2 in Human Vascular Smooth Muscle Cells. Front. Pharmacol. 2017, 7, 497. [Google Scholar] [CrossRef]
- Bang, E.; Kim, D.; Chung, H. Protease-activated receptor 2 induces ROS-mediated inflammation through Akt-mediated NF-κB and FoxO6 modulation during skin photoaging. Redox Biol. 2021, 44, 102022. [Google Scholar] [CrossRef]
- Subramaniam, S.; Ogoti, Y.; Hernandez, I.; Zogg, M.; Botros, F.; Burns, R.; DeRousse, J.; Dockendorff, C.; Mackman, N.; Antoniak, S.; et al. A thrombin-PAR1/2 feedback loop amplifies thromboinflammatory endothelial responses to the viral RNA analogue poly(I:C). Blood Adv. 2021, 5, 2760–2774. [Google Scholar] [CrossRef]
- Seo, Y.; Heo, Y.; Jo, S.; Park, S.; Lee, C.; Chang, J.; Jeon, D.; Kim, T.G.; Han, G.; Namkung, W. Novel positive allosteric modulator of protease-activated receptor 1 promotes skin wound healing in hairless mice. Br. J. Pharmacol. 2021, 178, 3414–3427. [Google Scholar] [CrossRef]
- Cheng, R.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.; Brown, G.; Cooke, R.; Dumelin, C.; Doré, A.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sekiguchi, F.; Hong, H.; Kawabata, A. PAR2 triggers IL-8 release via MEK/ERK and PI3-kinase/Akt pathways in GI epithelial cells. Biochem. Biophys. Res. Commun. 2008, 377, 622–626. [Google Scholar] [CrossRef]
- Pan, S.; Tao, K.; Guh, J.; Sun, H.; Huang, D.; Chang, Y.; Teng, C. The p38 mitogen-activated protein kinase pathway plays a critical role in PAR2-induced endothelial IL-8 production and leukocyte adhesion. Shock 2008, 30, 496–502. [Google Scholar] [CrossRef]
- Xu, W.; Andersen, H.; Whitmore, T.; Presnell, S.; Yee, D.; Ching, A.; Gilbert, T.; Davie, E.; Foster, D. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA 1998, 95, 6642–6646. [Google Scholar] [CrossRef]
- Böhm, S.; Khitin, L.; Grady, E.; Aponte, G.; Payan, D.; Bunnett, N. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 1996, 271, 22003–22016. [Google Scholar] [CrossRef]
- Her, J.; Lee, Y.; Kim, S.; Heo, G.; Choo, J.; Kim, Y.; Howe, C.; Rhee, S.; Yu, H.; Chung, H.; et al. Blockage of protease-activated receptor 2 exacerbates inflammation in high-fat environment partly through autophagy inhibition. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G30–G42. [Google Scholar] [CrossRef]
- Oakley, R.; Laporte, S.; Holt, J.; Caron, M.; Barak, L. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef]
- Oakley, R.; Laporte, S.; Holt, J.; Barak, L.; Caron, M. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J. Biol. Chem. 2001, 276, 19452–19460. [Google Scholar] [CrossRef]
- Gastel, J.V.; Hendrickx, J.; Leysen, H.; Santos-Otte, P.; Luttrell, L.; Martin, B.; Maudsley, S. β-Arrestin Based Receptor Signaling Paradigms: Potential Therapeutic Targets for Complex Age-Related Disorders. Front. Pharmacol. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Sharma, D.; Parameswaran, N. Multifaceted role of β-arrestins in inflammation and disease. Genes Immun. 2015, 16, 499–513. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, Y.; Yang, E.; Lee, Y.; Seo, Y.; Ryu, K.; Jeon, H.; Namkung, W. GB83, an Agonist of PAR2 with a Unique Mechanism of Action Distinct from Trypsin and PAR2-AP. Int. J. Mol. Sci. 2022, 23, 10631. https://doi.org/10.3390/ijms231810631
Heo Y, Yang E, Lee Y, Seo Y, Ryu K, Jeon H, Namkung W. GB83, an Agonist of PAR2 with a Unique Mechanism of Action Distinct from Trypsin and PAR2-AP. International Journal of Molecular Sciences. 2022; 23(18):10631. https://doi.org/10.3390/ijms231810631
Chicago/Turabian StyleHeo, Yunkyung, Eunhee Yang, Yechan Lee, Yohan Seo, Kunhi Ryu, Hyejin Jeon, and Wan Namkung. 2022. "GB83, an Agonist of PAR2 with a Unique Mechanism of Action Distinct from Trypsin and PAR2-AP" International Journal of Molecular Sciences 23, no. 18: 10631. https://doi.org/10.3390/ijms231810631