

NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NLRP3 Inflammasome
3. Pyroptosis
3.1. Caspase-1-Dependent Pathway
3.2. Caspase-1-Independent Pathway
4. NLRP3 Inflammasome/Pyroptosis in Diabetic Cardiomyopathy
4.1. Mechanism of NLRP3 Inflammasome/Pyroptosis in DCM
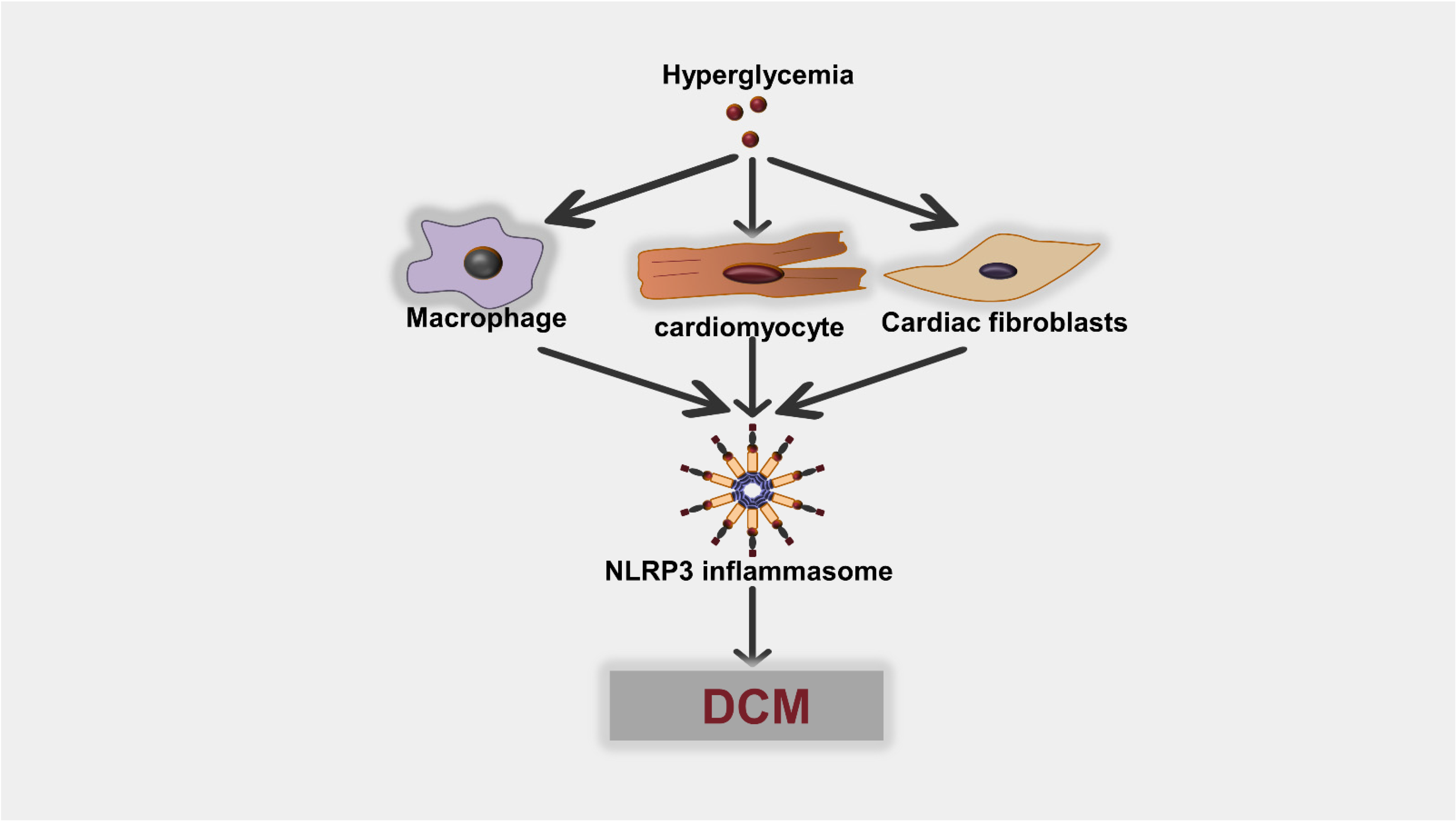
4.2. NLRP3 Inflammasome/Pyroptosis Activation in Different Cells in Diabetic Heart
4.3. The Role of NLRP3 Inflammasome/Pyroptosis on Cardiac Vasculature in DM/DCM
5. Potential Therapeutic Strategies for Targeting NLRP3 Inflammasome/Pyroptosis in DCM
5.1. Hypoglycemic Agents
5.2. Phytochemicals
5.3. Inhibiting Compounds
5.4. Non-Coding RNAs
5.5. Protein Molecules
6. Important Gaps in Knowledge in the Field
6.1. The Precise Relationship between Mitochondrial Dysfunction and NLRP3 Inflammasome
6.2. The Exact Regulatory Mechanism of NLRP3 Inflammasome/pyroptosis Activation in Macrophages in the Context of Diabetic Cardiomyopathy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACR | apoptosis repressor with caspase |
| AMI | acute myocardial infarction |
| AGEs | advanced glycation end products |
| AIM2 | absent in melanoma2-like receptors |
| AMPK | AMP-activated protein kinase |
| AS | atherosclerosis |
| ASC | apoptosis-associated speck-like protein containing |
| BTK | Bruton’s tyrosine kinase |
| CACR | caspase-1-associated circ RNA |
| CARD | caspase activation and recruitment domain |
| CFs | cardiac fibroblasts |
| CLRs | C-type lectin receptors |
| circ RNAs | circular RNAs |
| CMR | cardiac magnetic resonance |
| COX-2 | Cyclooxygenase 2 (COX-2) |
| CV | coriolus versicolor |
| CVDs | cardiovascular diseases |
| DAMP | damage- associated molecular patterns |
| DCM | diabetic cardiomyopathy |
| DPP4 | dipeptidyl peptidase4 |
| ELAVL1 | ELAV-like RNA-binding protein 1 |
| GBP | guanylate-binding protein |
| Gps | Gypenosides |
| GSDMD | Gasdermin D |
| GSDMD-CT | Gasdermin C-terminal domain |
| GSDMD-NT | Gasdermin N-terminal domain |
| G3BP1 | GTPase-activating protein (SH3 domain)-binding-protein 1 |
| GV | Glucose variability |
| HF | heart failure |
| HG | high glucose |
| H/R | Hypoxia/Reoxygenation |
| IL-1β | Interleukin-1β |
| IL-18 | Interleukin-18 |
| INF-γ | Interferon-γ |
| LGE | late gadolinium enhancement |
| LPS | Lipopolysaccharide |
| LRR | Leucine-rich repeat |
| ncRNAs | non-coding RNAs |
| NEK7 | NIMA-related kinase 7 |
| NF-κB | nuclear factors -κB |
| NLRP3 | NOD-like receptor pyrin domain containing 3 |
| oxLDL | oxidized low-density lipoprotein |
| PAMP | pathogens-associated molecular patterns |
| PCD | programmed cell death |
| PRR | pattern recognition receptor |
| PPARs | peroxisome proliferator-activated receptors |
| P2X7 | purinergic receptor P2X, ligand-gated ion channel 7 |
| RAMI | recurrent acute myocardial infarction |
| RLRs | retinoic acid-inducible gene (RIG)-I-like receptors |
| ROS | reactive oxygen species |
| SGLT2 | sodium-glucose cotransporter 2 |
| SGLT2i | sodium-glucose cotransporter 2 inhibitor |
| SIRT3 | sirtuin 3 |
| STZ | streptozotocin |
| T2D | type 2 diabetes |
| TLRs | toll-like receptors |
| TRX | thioredoxin |
| TXNIP | thioredoxin-interacting protein |
| USP19 | Ubiquitin-specific protease 19 |
| Vaspin | visceral adipose tissue-derived serine protease inhibitor |
References
- De Simone, G.; Devereux, R.B.; Chinali, M.; Lee, E.T.; Galloway, J.M.; Barac, A.; Panza, J.A.; Howard, B.V. Diabetes and incident heart failure in hypertensive and normotensive participants of the Strong Heart Study. J. Hypertens. 2010, 28, 353–360. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Maisch, B.; Alter, P.; Pankuweit, S. Diabetic cardiomyopathy—Fact or fiction? Herz 2011, 36, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C.; Calicchio, F.; Grassi, G.; Mancia, G. Diabetic cardiomyopathy: How can cardiac magnetic resonance help? Acta Diabetol. 2020, 57, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Kwong, R.Y.; Sattar, H.; Wu, H.; Vorobiof, G.; Gandla, V.; Steel, K.; Siu, S.; Brown, K.A. Incidence and prognostic implication of unrecognized myocardial scar characterized by cardiac magnetic resonance in diabetic patients without clinical evidence of myocardial infarction. Circulation 2008, 118, 1011–1020. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Jiaojiao, C.; Peng, W.; Yami, L.; Tingting, Z.; Jun, T.; Shiyuan, W.; Jinyan, X.; Dangheng, W.; Zhisheng, J.; et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J. Cell. Physiol. 2019, 234, 7475–7491. [Google Scholar] [CrossRef]
- Wu, P.; Chen, J.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-oxide promotes apoE(−/−) mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J. Cell. Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; He, F.; Cheng, C.; Xu, B.; Sheng, J. Uric acid aggravates myocardial ischemia-reperfusion injury via ROS/NLRP3 pyroptosis pathway. Biomed. Pharm. 2021, 133, 110990. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef]
- Li, W.; Chen, L.; Xiao, Y. Apigenin protects against ischemia-/hypoxia-induced myocardial injury by mediating pyroptosis and apoptosis. Vitr. Cell. Dev. Biol. Anim. 2020, 56, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Abdelaziz, D.H.; Mostafa, M.; Abdulrahman, B.A.; Grandhi, J.; Akhter, A.; Abu Khweek, A.; Aubert, D.F.; Valvano, M.A.; Wewers, M.D.; et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J. Immunol. 2012, 188, 3469–3477. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Georgin-Lavialle, S.; Grateau, G.; Amselem, S.; Giurgea, I.; Karabina, S.A. Inflammasome biology, molecular pathology and therapeutic implications. Pharm. Ther. 2018, 187, 133–149. [Google Scholar] [CrossRef]
- Wen, H.; Miao, E.A.; Ting, J.P. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 2013, 39, 432–441. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Lamkanfi, M. Snapshot of a Deadly Embrace: The Caspase-1-GSDMD Interface. Immunity 2020, 53, 6–8. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Chen, S.; Sun, B. Negative regulation of NLRP3 inflammasome signaling. Protein Cell 2013, 4, 251–258. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Gram, A.M.; Frenkel, J.; Ressing, M.E. Inflammasomes and viruses: Cellular defence versus viral offence. J. Gen. Viro.l 2012, 93, 2063–2075. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [Green Version]
- Kandli, M.; Feige, E.; Chen, A.; Kilfin, G.; Motro, B. Isolation and characterization of two evolutionarily conserved murine kinases (Nek6 and nek7) related to the fungal mitotic regulator, NIMA. Genomics 2000, 68, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Janicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Lamkanfi, M. Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb. Perspect. Biol. 2020, 12, a036392. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. N. Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef]
- Wilson, K.P.; Black, J.A.; Thomson, J.A.; Kim, E.E.; Griffith, J.P.; Navia, M.A.; Murcko, M.A.; Chambers, S.P.; Aldape, R.A.; Raybuck, S.A.; et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature 1994, 370, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 2019, 51, 43–49.e4. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Russo, H.M.; Rathkey, J.; Boyd-Tressler, A.; Katsnelson, M.A.; Abbott, D.W.; Dubyak, G.R. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J. Immunol. 2016, 197, 1353–1367. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef]
- Akhter, A.; Caution, K.; Abu Khweek, A.; Tazi, M.; Abdulrahman, B.A.; Abdelaziz, D.H.; Voss, O.H.; Doseff, A.I.; Hassan, H.; Azad, A.K.; et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 2012, 37, 35–47. [Google Scholar] [CrossRef]
- Knodler, L.A.; Crowley, S.M.; Sham, H.P.; Yang, H.; Wrande, M.; Ma, C.; Ernst, R.K.; Steele-Mortimer, O.; Celli, J.; Vallance, B.A. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef]
- Santos, J.C.; Dick, M.S.; Lagrange, B.; Degrandi, D.; Pfeffer, K.; Yamamoto, M.; Meunier, E.; Pelczar, P.; Henry, T.; Broz, P. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018, 37, e98089. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Devereux, R.B.; Roman, M.J.; Paranicas, M.; O’Grady, M.J.; Lee, E.T.; Welty, T.K.; Fabsitz, R.R.; Robbins, D.; Rhoades, E.R.; Howard, B.V. Impact of diabetes on cardiac structure and function: The strong heart study. Circulation 2000, 101, 2271–2276. [Google Scholar] [CrossRef]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen remodelling in myocardia of patients with diabetes. J. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K.; Kiriazis, H.; Du, X.J.; Love, J.E.; Gray, S.P.; Jandeleit-Dahm, K.A.; McMullen, J.R.; Ritchie, R.H. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic. Biol. Med. 2013, 60, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.M.; Mouton, S.; Sebti, Y.; et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hua, Y.; Li, X.; Arslan, I.M.; Zhang, W.; Meng, G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front Pharm. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Parim, B.; Sathibabu Uddandrao, V.V.; Saravanan, G. Diabetic cardiomyopathy: Molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart Fail. Rev. 2018, 24, 279–299. [Google Scholar] [CrossRef]
- Bolivar, B.E.; Vogel, T.P.; Bouchier-Hayes, L. Inflammatory caspase regulation: Maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019, 286, 2628–2644. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int. J. Biol. Sci. 2019, 15, 1345–1357. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.-F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.-F.; Ferré, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2017, 61, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, K.R.; Lord, C.K.; West, T.A.; Stewart, J.A., Jr. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS ONE 2013, 8, e72080. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, E.C.; Bradshaw, A.D.; Zile, M.R.; Spinale, F.G. Myocardial fibroblast-matrix interactions and potential therapeutic targets. J. Mol. Cell. Cardiol. 2014, 70, 92–99. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Zhao, X.D.; Shi, K.H.; Ding, X.S.; Tao, H. MiR-21-3p triggers cardiac fibroblasts pyroptosis in diabetic cardiac fibrosis via inhibiting androgen receptor. Exp. Cell Res. 2021, 399, 112464. [Google Scholar] [CrossRef]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.Y. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid. Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef]
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836. [Google Scholar] [CrossRef]
- Bardini, G.; Dicembrini, I.; Cresci, B.; Rotella, C.M. Inflammation markers and metabolic characteristics of subjects with 1-h plasma glucose levels. Diabetes Care 2010, 33, 411–413. [Google Scholar] [CrossRef]
- Muller, S.; Martin, S.; Koenig, W.; Hanifi-Moghaddam, P.; Rathmann, W.; Haastert, B.; Giani, G.; Illig, T.; Thorand, B.; Kolb, H. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia 2002, 45, 805–812. [Google Scholar] [CrossRef]
- Urbina, P.; Singla, D.K. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H762–H772. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.B.; Wei, G.S.; Li, F.X.; Guo, W.J.; Hong, P.; Weng, Y.Q.; Zhang, Q.Q.; Xu, S.Y.; Liang, W.B.; You, Z.J.; et al. Macrophage-NLRP3 Inflammasome Activation Exacerbates Cardiac Dysfunction after Ischemic Stroke in a Mouse Model of Diabetes. Neurosci. Bull. 2020, 36, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Purvis, G.S.D.; Collino, M.; Aranda-Tavio, H.; Chiazza, F.; O’Riordan, C.E.; Zeboudj, L.; Mohammad, S.; Collotta, D.; Verta, R.; Guisot, N.E.S.; et al. Inhibition of Bruton’s TK regulates macrophage NF-kappaB and NLRP3 inflammasome activation in metabolic inflammation. Br. J. Pharm. 2020, 177, 4416–4432. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Cellular and molecular mechanisms of vascular injury in diabetes—Part II: Cellular mechanisms and therapeutic targets. Vasc. Pharm. 2011, 54, 75–79. [Google Scholar] [CrossRef]
- Correa, R.; Silva, L.F.F.; Ribeiro, D.J.S.; Almeida, R.D.N.; Santos, I.O.; Correa, L.H.; de Sant’Ana, L.P.; Assuncao, L.S.; Bozza, P.T.; Magalhaes, K.G. Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front. Immunol. 2019, 10, 2927. [Google Scholar] [CrossRef]
- Zeng, Z.; Zheng, Q.; Chen, J.; Tan, X.; Li, Q.; Ding, L.; Zhang, R.; Lin, X. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 2020, 393, 112108. [Google Scholar] [CrossRef]
- Burleigh, M.E.; Babaev, V.R.; Yancey, P.G.; Major, A.S.; McCaleb, J.L.; Oates, J.A.; Morrow, J.D.; Fazio, S.; Linton, M.F. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J. Mol. Cell. Cardiol. 2005, 39, 443–452. [Google Scholar] [CrossRef]
- Hua, K.F.; Chou, J.C.; Ka, S.M.; Tasi, Y.L.; Chen, A.; Wu, S.H.; Chiu, H.W.; Wong, W.T.; Wang, Y.F.; Tsai, C.L.; et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1beta production. J. Cell. Physiol. 2015, 230, 863–874. [Google Scholar] [CrossRef]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, R.M.; Nesto, R.W. Acute myocardial infarction in the diabetic patient: Pathophysiology, clinical course and prognosis. J. Am. Coll. Cardiol. 1992, 20, 736–744. [Google Scholar] [CrossRef]
- van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Song, M.Y.; Li, C.F.; Zhao, J.Q. shRNA interference of NLRP3 inflammasome alleviate ischemia reperfusion-induced myocardial damage through autophagy activation. Biochem. Biophys. Res. Commun. 2017, 494, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Monnier, L.; Owens, D. Glycaemic variability in diabetes: Clinical and therapeutic implications. Lancet Diabetes Endocrinol. 2019, 7, 221–230. [Google Scholar] [CrossRef]
- Xia, J.; Zhang, J.; Chang, J.; Tian, Y.; Li, J.; Zhang, B.; Zeng, X.; Yin, C. The effects of glycaemic variability on intimal hyperplasia and plaque stability after stenting via autophagy-mediated G3BP1/NLRP3 inflammasome. Ann Transl Med 2020, 8, 1388. [Google Scholar] [CrossRef]
- Flory, J.; Lipska, K. Metformin in 2019. JAMA 2019, 321, 1926–1927. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE(−/−) mice. Biomed. Pharm. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Fei, Q.; Ma, H.; Zou, J.; Wang, W.; Zhu, L.; Deng, H.; Meng, M.; Tan, S.; Zhang, H.; Xiao, X.; et al. Metformin protects against ischaemic myocardial injury by alleviating autophagy-ROS-NLRP3-mediated inflammatory response in macrophages. J. Mol. Cell. Cardiol. 2020, 145, 1–13. [Google Scholar] [CrossRef]
- Ferrannini, E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Jhund, P.S.; Docherty, K.F.; Diez, M.; Petrie, M.C.; Verma, S.; Nicolau, J.C.; Merkely, B.; Kitakaze, M.; DeMets, D.L.; et al. Effects of Dapagliflozin on Symptoms, Function, and Quality of Life in Patients With Heart Failure and Reduced Ejection Fraction: Results From the DAPA-HF Trial. Circulation 2020, 141, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.M.; Gandy, S.; McCrimmon, R.; Houston, J.G.; Struthers, A.D.; Lang, C.C. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: The DAPA-LVH trial. Eur. Heart J. 2020, 41, 3421–3432. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tran, D.; Yang, H.C.; Nylander, S.; Birnbaum, Y.; Ye, Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: An AMPK-mTOR Interplay. Cardiovasc. Drugs Ther. 2020, 34, 443–461. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.S.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated With Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, e006277. [Google Scholar] [CrossRef]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 20, 150. [Google Scholar] [CrossRef]
- Gordon, M.; Meagher, P.; Connelly, K.A. Effect of Empagliflozin and Liraglutide on the Nucleotide-Binding and Oligomerization Domain-Like Receptor Family Pyrin Domain-Containing 3 Inflammasome in a Rodent Model of Type 2 Diabetes Mellitus. Can. J. Diabetes 2021, 45, 553–556. [Google Scholar] [CrossRef]
- Lee, T.W.; Bai, K.J.; Lee, T.I.; Chao, T.F.; Kao, Y.H.; Chen, Y.J. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 2017, 24, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-C.; Wu, C.-H.; Lin, T.-C.; Cheng, Y.-N.; Chang, C.-S.; Lee, K.-T.; Tsai, P.-J.; Tsai, Y.-S. Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics 2021, 11, 2424–2441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Zong, B.; Yuan, H.; Wang, Z.; Wei, Y.; Wang, X.; Liu, G.; Zhang, J.; Li, S.; et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J. Cell. Mol. Med. 2018, 22, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Y.; Jin, Y.; Chen, Y.; Tian, L.; He, W. Synergistic cardioptotection by tilianin and syringin in diabetic cardiomyopathy involves interaction of TLR4/NF-kappaB/NLRP3 and PGC1a/SIRT3 pathways. Int. Immunopharmacol. 2021, 96, 107728. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Li, Y.; Zhao, Y.; Xiong, F.; Liu, Y.; Xue, H.; Yang, Z.; Ni, S.; Sahil, A.; et al. Coriolus versicolor alleviates diabetic cardiomyopathy by inhibiting cardiac fibrosis and NLRP3 inflammasome activation. Phytother. Res. 2019, 33, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, R.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in Patients With Acute Coronary Syndrome: The Australian COPS Randomized Clinical Trial. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef]
- Martinez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Marques-da-Silva, C.; Chaves, M.M.; Castro, N.G.; Coutinho-Silva, R.; Guimaraes, M.Z. Colchicine inhibits cationic dye uptake induced by ATP in P2X2 and P2X7 receptor-expressing cells: Implications for its therapeutic action. Br. J. Pharm. 2011, 163, 912–926. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, K.; Zhou, Y.; Wang, H.; Zhou, Y.; Liu, S.; Nie, Y.; Li, Y. NLRP3 inflammasome inhibitor CY-09 reduces hepatic steatosis in experimental NAFLD mice. Biochem. Biophys. Res. Commun. 2021, 534, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef]
- Sharma, A.; Choi, J.S.Y.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E.; et al. Specific NLRP3 Inhibition Protects Against Diabetes-Associated Atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, W.; Ma, W.; Zhou, X.; Quan, Z.; Li, G.; Liu, D.; Zhang, Q.; Han, D.; Gao, B.; et al. (18)F-FDG PET imaging-monitored anti-inflammatory therapy for acute myocardial infarction: Exploring the role of MCC950 in murine model. J. Nucl. Cardiol. 2021, 28, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, X.; Chen, X.; Yang, S.; Chen, H. Inhibition of miR-223 attenuates the NLRP3 inflammasome activation, fibrosis, and apoptosis in diabetic cardiomyopathy. Life Sci. 2020, 256, 117980. [Google Scholar] [CrossRef]
- Li, X.; Du, N.; Zhang, Q.; Li, J.; Chen, X.; Liu, X.; Hu, Y.; Qin, W.; Shen, N.; Xu, C.; et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014, 5, e1479. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Lv, J.; Wang, Y.; Che, H.; Chen, X.; Jiang, Y.; Li, A.; Sun, X.; Yue, E.; et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018, 9, 1000. [Google Scholar] [CrossRef] [Green Version]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/Smads signaling in diabetic cardiomyopathy. FASEB J. 2020, 34, 5282–5298. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, A.; Qin, Y.; Che, H.; Wang, Y.; Lv, J.; Li, Y.; Li, H.; Yue, E.; Ding, X.; et al. A Novel Circular RNA Mediates Pyroptosis of Diabetic Cardiomyopathy by Functioning as a Competing Endogenous RNA. Mol. Ther. Nucleic Acids 2019, 17, 636–643. [Google Scholar] [CrossRef]
- Li, X.; Ke, X.; Li, Z.; Li, B. Vaspin prevents myocardial injury in rats model of diabetic cardiomyopathy by enhancing autophagy and inhibiting inflammation. Biochem. Biophys. Res. Commun. 2019, 514, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; Li, B.; Zhu, X.; Lai, X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sci. 2019, 234, 116773. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ding, Y.; Dai, G.L.; Zhang, Y.; Xu, M.T.; Shen, J.R.; Chen, T.T.; Chen, Y.; Meng, G.L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by beta-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, M.; Zhang, Y.; Guo, H.; Ding, W.; Sun, T. Nlrp3 Deficiency Alleviates Angiotensin II-Induced Cardiomyopathy by Inhibiting Mitochondrial Dysfunction. Oxidative Med. Cell. Longev. 2021, 2021, 6679100. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Li, C.; Jin, Y.; Wei, S.; Sun, Y.; Jiang, L.; Zhu, Q.; Farmer, D.G.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Ke, B. Hippo Signaling Controls NLR Family Pyrin Domain Containing 3 Activation and Governs Immunoregulation of Mesenchymal Stem Cells in Mouse Liver Injury. Hepatology 2019, 70, 1714–1731. [Google Scholar] [CrossRef]
- Liu, T.; Wang, L.; Liang, P.; Wang, X.; Liu, Y.; Cai, J.; She, Y.; Wang, D.; Wang, Z.; Guo, Z.; et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell. Mol. Immunol. 2021, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ai, C.; Bai, M.; Niu, J.; Zhang, Z. NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 10632. https://doi.org/10.3390/ijms231810632
Zhang L, Ai C, Bai M, Niu J, Zhang Z. NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2022; 23(18):10632. https://doi.org/10.3390/ijms231810632
Chicago/Turabian StyleZhang, Lixia, Chenchen Ai, Ming Bai, Jinglei Niu, and Zheng Zhang. 2022. "NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy" International Journal of Molecular Sciences 23, no. 18: 10632. https://doi.org/10.3390/ijms231810632
APA StyleZhang, L., Ai, C., Bai, M., Niu, J., & Zhang, Z. (2022). NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy. International Journal of Molecular Sciences, 23(18), 10632. https://doi.org/10.3390/ijms231810632