


Involvement of Inflammation and Its Resolution in Disease and Therapeutics
 and
and
Abstract
:1. Introduction
2. Inflammation
3. Inflammation and Associated Diseases
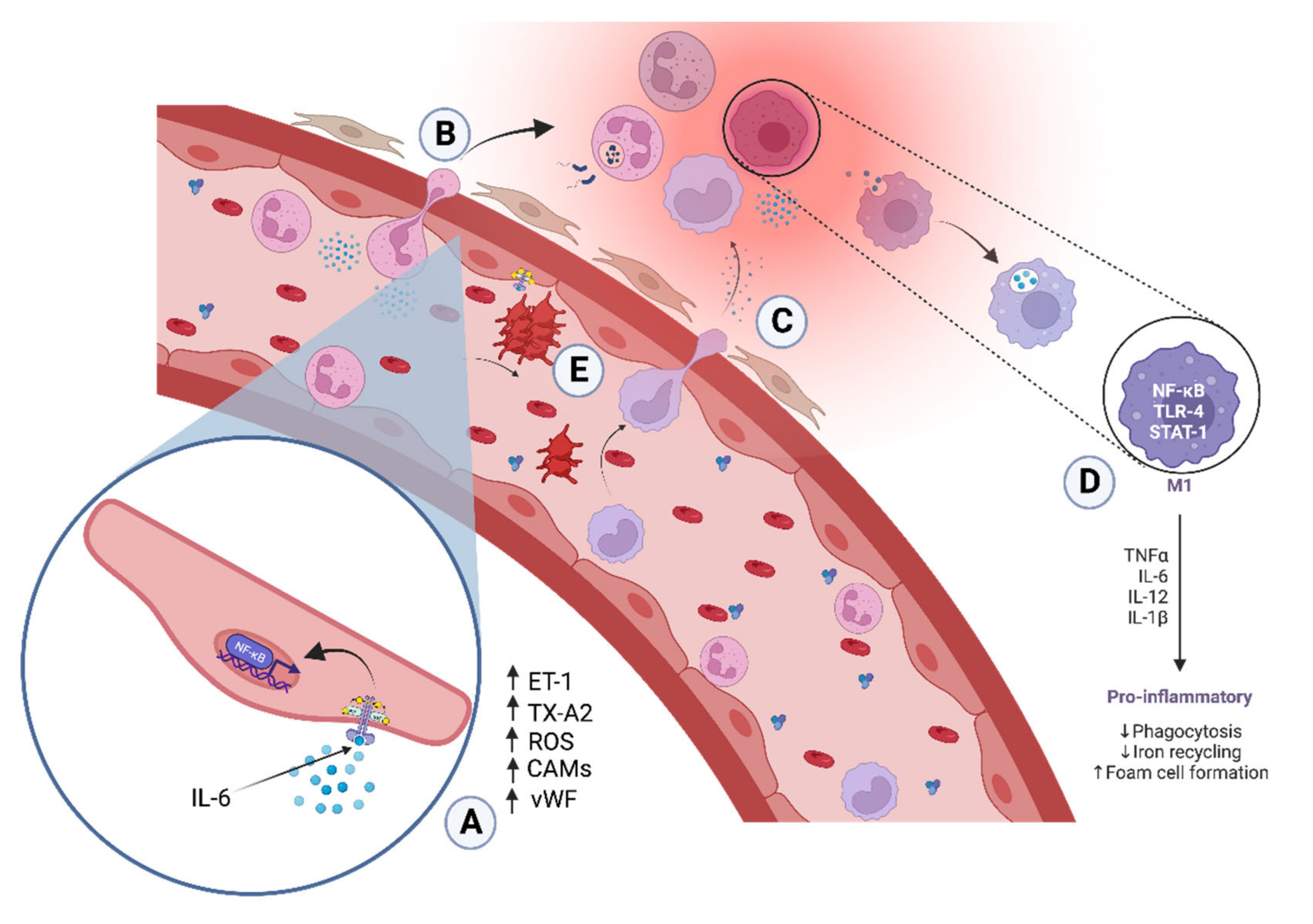
3.1. Endothelial Inflammation in Atherosclerosis
3.2. Pathogens-Anduced Inflammation
3.2.1. Bacterial Sepsis
3.2.2. Viruses and Immune Response
3.2.3. Parasitic Infection in Chagas Disease
3.3. Adjuvants-Induced Inflammation
Uncontrolled Inflammation Caused by Adjuvants in Vaccines
3.4. Wound Healing
3.4.1. Phases of Wound Healing
3.4.2. Chronic Wounds
4. Therapeutics
4.1. Conventional Therapies
4.2. Advanced Therapies for Wound Healing
4.2.1. Cell Therapy
4.2.2. Tissue Engineering
4.3. Resolution of Inflammation as a Novel Therapeutic Target
4.3.1. Specialized Pro-Resolving Mediators Mark the Dawn of the Resolution Pharmacology
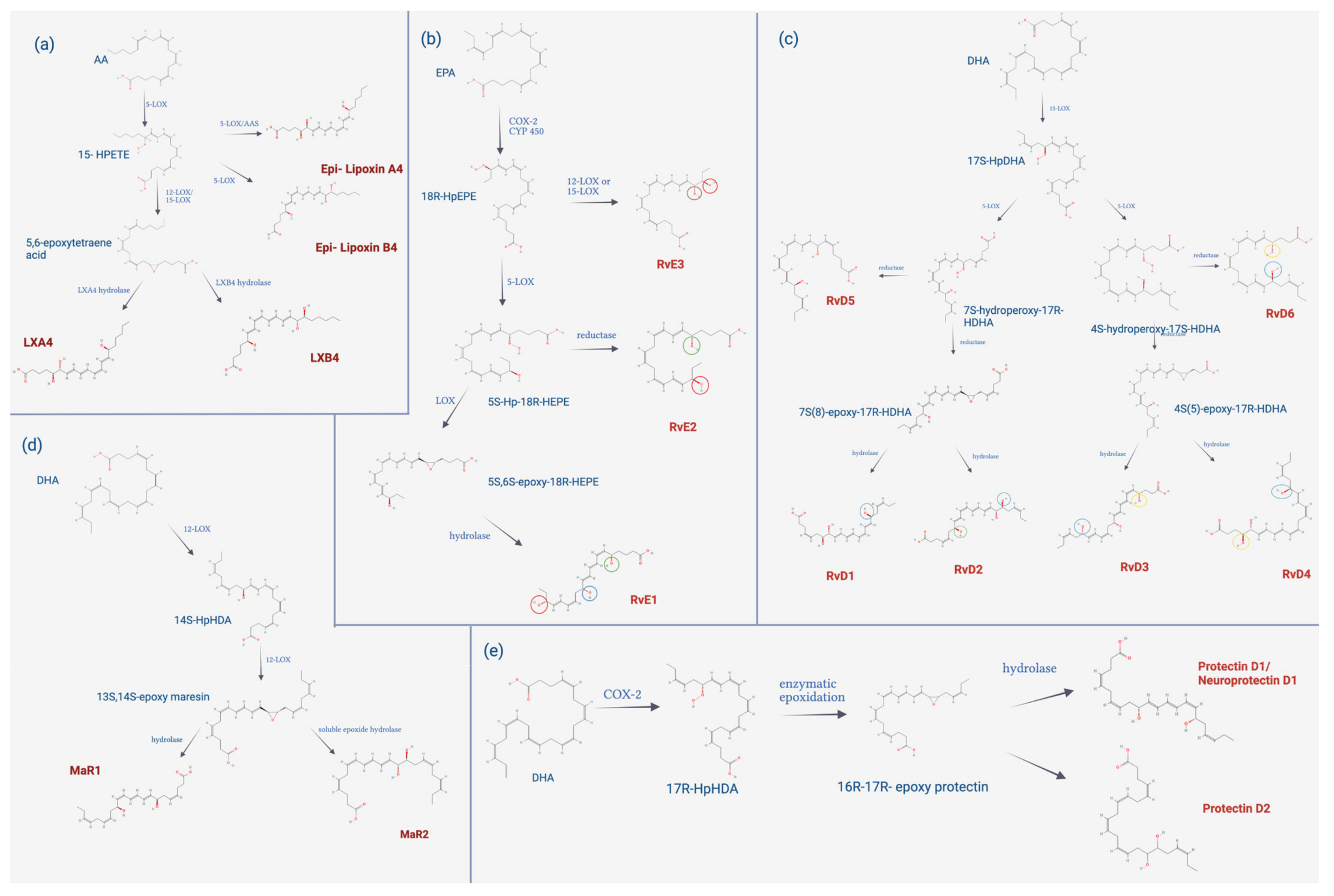
4.3.2. Biosynthesis of SPMs
4.3.3. Biological Functions of SPMs
4.3.4. Limitations and Future Directions in Resolution Pharmacology
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 12-LOX | 12-Lipoxygenase |
| 13R,14S-diHDHA | 13R,14S-dihydroxy-docosahexaenoic acid |
| 14S–HpDHA | 14S-hydroperoxy-docosahexaenoic acid |
| 15-epi-LXA4 | 15-epi-lipoxin A4 |
| 15-HPETE | 15-hydroperoxy-eicosatetraenoic acid |
| 15-LOX | 15-lipoxygenase |
| 15-PGDH | 15-prostaglandin dehydrogenase |
| 17-oxo-RvD1 | 17-oxoresolvin D1 |
| 17S-HpDHA | 17S-hydroperoxy-docosahexaenoic acid |
| 18R-HEPE | 18R-hydroxy-eicosapentaenoic acid |
| 18R-HpEPE | 18R-hydroxyperoxy-eicosapentaenoic acid |
| 5-LOX | 5-lipoxygenase |
| 8-oxo-RvD1 | 8-oxoresolvin D1 |
| AA | arachidonic acid |
| APC | antigen-presenting cell |
| ARG | arginase |
| AT-L | aspirin-triggered analogs |
| BDA-RvD1 | benzo-diacetylene-17R-RvD1 |
| benzo-LXA4 | benzo-lipoxin A4 |
| bFGF | basic fibroblast growth factor |
| BLT1 | leukotriene B4 receptor 1 |
| CAD | coronary artery disease |
| cAMP | cyclic adenosine monophosphate |
| CAMs | cell adhesion molecules |
| CANTOS | canakinumab anti-inflammatory thrombosis outcomes study |
| CCL2 | chemokine C-C motif ligand 2 |
| CCL20 | chemokine C-C motif ligand 20 |
| CCL3 | chemokine C-C motif ligand 3 |
| CCL4 | chemokine C-C motif ligand 4 |
| CCL8 | chemokine C-C motif ligand 8 |
| CCR5 | receptor 5 chemokine cysteine-cysteine cysteine |
| CD105 | cluster of differentiation 105 |
| CD11b | cluster of differentiation 11b |
| CD14 | cluster of differentiation 14 |
| CD19 | cluster of differentiation 19 |
| CD34 | cluster of differentiation 34 |
| CD73 | cluster of differentiation 73 |
| CD79α | cluster of differentiation 79 α |
| CD80 | cluster of differentiation 80 |
| CD83 | cluster of differentiation 83 |
| CD86 | cluster of differentiation 86 |
| CD90 | cluster of differentiation 90 |
| ChemR23 | chemerin receptor 23 |
| CIRT | cardiovascular Inflammation Reduction Trial |
| COX | cyclooxygenase |
| COX-2 | cyclooxygenase-2 |
| CRP | C-reactive protein |
| CRS | Cytokine Release Syndrome |
| CVDs | cardiovascular diseases |
| CXCL1 | chemokine C-X-C motif ligand-1 |
| CXCL2 | chemokine C-X-C motif ligand-2 |
| CXCL5 | chemokine C-X-C motif ligand-5 |
| CYP | cytochrome |
| CYP1A | cytochrome P450 1A |
| CYP2B1/2 | cytochrome P450 2B1/2 |
| CYP3A4 | cytochrome P450 3A4 |
| DAMPs | damage associated molecular patterns |
| DCs | dendritic cells |
| DHA | docosahexaenoic acid |
| E-CAMs | extracellular matrix adhesion molecules |
| ECM | extracellular matrix |
| EOR | eicosanoid oxidoreductase |
| EPA | eicosapentaenoic acid |
| ET-1 | endothelin-1 |
| FDA | food and drug administration |
| FGF | fibroblast growth factor |
| FPR2/ALX | formyl peptide receptor 2 |
| FPR2/ALX | formyl peptide receptor 2 |
| GPR18 | G protein-coupled receptor 18 |
| GPR32 | G protein-couple receptor 32 |
| GPR37 | G protein-coupled receptor 37 |
| HLA DR | human leukocyte antigen—DR isotype |
| IBS | innate immune system |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFN | interferon |
| IFN-α/β | interferons alpha/beta |
| IFN-β | interferon β |
| IFN- γ | interferon γ |
| IL-1 | interleukin-1 |
| IL-1β | interleukin-1 β |
| IL-10 | interleukin-10 |
| IL-12 | interleukin-12 |
| IL-13 | Interleukin-13 |
| IL-17β | interleukin- 17 β |
| IL-2 | interleukin-2 |
| IL-23 | interleukin-23 |
| IL-6 | interleukin-6 |
| IRF7 | interferon regulatory factor 7 |
| LDL | low density lipoprotein |
| LGR6 | G protein-coupled receptor 6 |
| LOX | lipoxygenase |
| LPS | lipopolysaccharide |
| LXA4 | lipoxin A4 |
| LXB4 | lipoxin B4 |
| LXs | lipoxins |
| MAIT | mucosal Associated Innate T cells |
| MaR1 | maresin 1 |
| MaR2 | maresine 2 |
| MCP-1 | monocyte chemoattractant protein-1 |
| MHC | major histocompatibility molecules |
| miRNA | microRNA |
| MPL | monophosphoryl lipid A |
| MSCs | mesenchymal stem cells |
| MyD88/RAK/TRAF6 | myeloid differentiation factor 88/IL-1 receptor-associated kinase 1/tumor necrosis factor receptor-associated factor 6 |
| MyD88/TIRAP | myeloid differentiation factor 88/domain-containing adaptor protein |
| NF-κB | nuclear factor kappa B |
| NLRs | nod-like receptors |
| NO | nitric oxide |
| NOTCH-1 | neurogenic locus notch homolog protein 1 |
| NPD1 | neuroprotectin D1 |
| PAMPs | pathogen associated molecular patterns |
| PD1 | protectin D1 |
| PDGF | platelet-derived growth factor |
| PECAM-1 | platelet endothelial cell adhesion molecule-1 |
| PGDH | prostaglandin dehydrogenase |
| PGI2 | prostacyclin |
| PMN | polymorphonuclear neutrophils |
| PRRs | pattern recognition receptors |
| ROS | reactive oxygen species |
| RvD1 | resolvin D1 |
| RvD1-6 | D-series resolvins |
| RvD2 | resolvin D2 |
| RvD4 | resolvin D4 |
| RvD5 | resolvin D5 |
| RvE1 | resolvin E1 |
| RvE2 | resolvin E2 |
| RvE3 | resolvin E3 |
| SIE | specific immune system |
| SPM | specialized pro-resolving |
| T cruzi | Trypanosoma cruzi |
| TGF-β | transforming growth factor β |
| TGF-β3 | transforming growth factor β3 |
| TLR2 | toll-like receptor 2 |
| TLR4 | toll-like receptor 4 |
| TLRs | toll-like receptors |
| TNFα | tumor necrosis factor alpha |
| TRIF/TRAM | TIR-domain-containing adapter-inducing interferon-β/TRIF-related adaptor molecule |
| TSG-6 | tumor necrosis factor alfa stimulated gene 6 |
| TXA2 | thromboxane A2 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
| VSMCs | vascular smooth muscle cells |
References
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc. Med. 2022, 23, 73. [Google Scholar] [CrossRef]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pathogen recognition by innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Zhong, J.; Shi, G. Editorial: Regulation of Inflammation in Chronic Disease. Front. Immunol. 2019, 10, 737. [Google Scholar] [CrossRef]
- McCracken, J.L.; Veeranki, S.P.; Ameredes, B.T.; Calhoun, W.J. Diagnosis and management of asthma in adults a review. JAMA 2017, 318, 279–290. [Google Scholar] [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St.Clair, E.W.; Arayssi, T.; Carandang, K. American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2021, 73, 924–939. [Google Scholar] [CrossRef]
- Berliner, J.A.; Leitinger, N.; Tsimikas, S. The role of oxidized phospholipids in atherosclerosis. J. Lipid Res. 2009, 50 (Suppl.), S207–S212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grebe, A.; Latz, E. Cholesterol Crystals and Inflammation. Curr. Rheumatol. Rep. 2013, 15, 313. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M. Inflammation and Atherothrombosis: From Population Biology and Bench Research to Clinical Practice. J. Am. Coll. Cardiol. 2006, 48 (Suppl. 9), A33–A46. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Bezsonov, E.; Sobenin, I.; Orekhov, A. Immunopathology of Atherosclerosis and Related Diseases: Focus on Molecular Biology. Int. J. Mol. Sci. 2021, 22, 4080. [Google Scholar] [CrossRef]
- Muto, A.; Model, L.; Ziegler, K.; Eghbalieh, S.D.D.; Dardik, A. Mechanisms of vein graft adaptation to the arterial circulation—Insights into the neointimal algorithm and management strategies. Circ. J. 2010, 74, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Mottola, G.; Schaller, M.; Upchurch, G.R.; Conte, M.S. Resolution of vascular injury: Specialized lipid mediators and their evolving therapeutic implications. Mol. Aspects Med. 2017, 58, 72–82. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef]
- Tang, X.D.; Ji, T.T.; Dong, J.R.; Feng, H.; Chen, F.Q.; Chen, X.; Zhao, H.Y.; Chen, D.K.; Ma, W.T. Pathogenesis and treatment of cytokine storm induced by infectious diseases. Int. J. Mol. Sci. 2021, 22, 13009. [Google Scholar] [CrossRef]
- Reyes, M.; Filbin, M.R.; Bhattacharyya, R.P.; Billman, K.; Eisenhaure, T.; Hung, D.T.; Levy, B.D.; Baron, R.M.; Blainey, P.C.; Goldberg, M.B.; et al. An immune-cell signature of bacterial sepsis. Nat. Med. 2020, 26, 333–340. [Google Scholar] [CrossRef]
- Cavaillon, J.-M. Exotoxins and endotoxins: Inducers of inflammatory cytokines. Toxicon 2018, 149, 45–53. [Google Scholar] [CrossRef]
- Drewry, A.M.; Samra, N.; Skrupky, L.P.; Fuller, B.; Compton, S.M.; Hotchkiss, R.S. Persistent Lymphopenia After Diagnosis of Sepsis Predicts Mortality. Shock 2014, 42, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, M.; Sudhoff, L.; Crucian, B.; Pagel, J.-I.; Sams, C.; Strewe, C.; Guo, A.; Schelling, G.; Briegel, J.; Kaufmann, I.; et al. Early immune anergy towards recall antigens and mitogens in patients at onset of septic shock. Sci. Rep. 2018, 8, 1754. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Ogura, H.; Shimizu, K.; Ikeda, M.; Hirose, T.; Matsuura, H.; Kang, S.; Takahashi, K.; Tanaka, T.; Shimazu, T. The clinical importance of a cytokine network in the acute phase of sepsis. Sci. Rep. 2018, 8, 13995. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232. [Google Scholar] [CrossRef]
- Florindo, H.F.; Kleiner, R.; Vaskovich-koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-mediated approaches against COVID-19. Nature 2020, 15, 630–645. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The Inflammasome in Times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Alexandris, N.; Konstantinou, E.; Mesiakaris, K.; Zanidis, C.; Farsalinos, K.; Poulas, K. Imiquimod—A toll like receptor 7 agonist—Is an ideal option for management of COVID 19. Environ. Res. 2020, 188, 109858. [Google Scholar] [CrossRef]
- Dhar, S.K.; Vishnupriyan, K.; Damodar, S.; Gujar, S.; Das, M. IL-6 and IL-10 as predictors of disease severity in COVID-19 patients: Results from meta-analysis and regression. Heliyon 2021, 7, e06155. [Google Scholar] [CrossRef] [PubMed]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in Covid-19: A systematic review and meta-analysis. Reviews in Medical. Virology 2020, 30, 6. [Google Scholar]
- Gubernatorova, E.O.; Gorshkova, E.A.; Polinova, A.I.; Drutskaya, M.S. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kurioka, A.; Ussher, J.E.; Cosgrove, C.; Clough, C.; Fergusson, J.R.; Smith, K.; Kang, Y.H.; Walker, L.J.; Hansen, T.H.; Willberg, C.B.; et al. MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol. 2015, 8, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Meermeier, E.W.; Harriff, M.J.; Karamooz, E.; Lewinsohn, D.M. MAIT cells and microbial immunity. Immunol. Cell Biol. 2018, 96, 607–617. [Google Scholar] [CrossRef]
- Cosgrove, C.; Ussher, J.E.; Rauch, A.; Ga, K.; Kurioka, A.; Hu, M.H.; Adelmann, K.; Kang, Y.H.; Fergusson, J.R.; Simmonds, P.; et al. Early and nonreversible decrease of CD161++/MAIT cells in HIV infection. Blood 2015, 121, 951–962. [Google Scholar] [CrossRef]
- Parrot, T.; Gorin, J.B.; Ponzetta, A.; Maleki, K.T.; Kammann, T.; Emgård, J.; Perez-Potti, A.; Sekine, T.; Rivera-Ballesteros, O.; Karolinska COVID-19 Study Group; et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol. 2020, 5, eabe1670. [Google Scholar] [CrossRef]
- Yang, Q.; Wen, Y.; Qi, F.; Gao, X.; Chen, W.; Xu, G.; Wei, C.; Wang, H.; Tang, X.; Lin, J.; et al. Suppressive Monocytes Impair MAIT Cells Response via IL-10 in Patients with Severe COVID-19. J. Immunol. 2021, 207, 1848–1856. [Google Scholar] [CrossRef]
- Andrade, D.; Serra, R.; Svensjö, E.; Lima, A.P.; Ramos, E.S., Jr.; Fortes, F.S.; Morandini, A.C.; Morandi, V.; Soeiro, M.D.N.; Tanowitz, H.B.; et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: A converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 2012, 165, 1333–1347. [Google Scholar] [CrossRef]
- Molina-Berríos, A.; Campos-Estrada, C.; Lapier, M.; Duaso, J.; Kemmerling, U.; Galanti, N.; Leiva, M.; Ferreira, J.; López-Muñoz, R.; Maya, J.D. Benznidazole prevents endothelial damage in an experimental model of Chagas disease. Acta Trop. 2013, 127, 6–13. [Google Scholar] [CrossRef]
- Campos-Estrada, C.; Liempi, A.; González-Herrera, F.; Lapier, M.; Kemmerling, U.; Pesce, B.; Ferreira, J.; López-Muñoz, R.; Maya, J.D. Simvastatin and Benznida-zole-Mediated Prevention of Trypanosoma cruzi-Induced Endothelial Activation: Role of 15-epi-lipoxin A4 in the Action of Simvastatin. PLoS Negl. Trop. Dis. 2015, 9, e0003770. [Google Scholar] [CrossRef] [PubMed]
- Estrada, C.C.; González-Herrera, F.; Greif, G.; Carillo, I.; Guzmán-Rivera, D.; Liempi, A.; Robello, C.; Kemmerling, U.; Castillo, C.; Maya, J.D. Notch receptor expression in Trypanosoma cruzi -infected human umbilical vein endothelial cells treated with benznidazole or simvastatin revealed by microarray analysis. Cell Biol. Int. 2020, 44, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.H.; Kim, H.; Lee, J.; Kwon, H.H.; Park, G.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, I.; Rabelo, R.A.N.; Barbosa, C.; Rates, M.; Fuentes-Retamal, S.; González-Herrera, F.; Guzmán-Rivera, D.; Quintero, H.; Kemmerling, U.; Castillo, C.; et al. Aspirin-triggered resolvin D1 reduces parasitic cardiac load by decreasing inflammation in a murine model of early chronic Chagas disease. PLoS Neglected Trop. Dis. 2021, 15, e0009978. [Google Scholar] [CrossRef]
- Aguilar, J.C.; Rodríguez, E.G. Vaccine adjuvants revisited. Vaccine 2007, 25, 3752–3762. [Google Scholar] [CrossRef]
- Garçon, N.; Leroux-Roels, G.; Cheng, W.F. Understanding Modern Vaccines: Perspectives in Vaccinology. Perspect Vaccinol. 2011, 1, 89–113. [Google Scholar] [CrossRef]
- Petrovsky, N. Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Plotkin, S.A.; Black, S.; Coffman, R.L. Assessing the Safety of Adjuvanted Vaccines. Sci. Transl. Med. 2011, 3, 93rv2. [Google Scholar] [CrossRef]
- Batista-Duharte, A.; Lindblad, E.B.; Oviedo-Orta, E. Progress in understanding adjuvant immunotoxicity mechanisms. Toxicol. Lett. 2011, 203, 97–105. [Google Scholar] [CrossRef]
- McKinney, B.A.; Reif, D.M.; Rock, M.T.; Edwards, K.M.; Kingsmore, S.; Moore, J.H.; Crowe, J.J.E. Cytokine Expression Patterns Associated with Systemic Adverse Events following Smallpox Immunization. J. Infect. Dis. 2006, 194, 444–453. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, E.; D’Oro, U.; Wack, A. Immunology of TLR-independent vaccine adjuvants. Curr. Opin. Immunol. 2009, 21, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Friede, M.; Reed, S.G.; Ireton, G.C. Synthetic and Natural TLR4 Agonists as Safe and Effective Vaccine Adjuvants. In Endotoxins: Structure, Function and Recognition; Springer: Dordecht, The Netherlands, 2010; Volume 53, pp. 303–321. [Google Scholar]
- Alemzadeh, E.; Oryan, A.; Mohammadi, A.A. Hyaluronic acid hydrogel loaded by adipose stem cells enhances wound healing by mod-ulating IL-1β, TGF-β1, and bFGF in burn wound model in rat. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Campos-Estrada, C.; Riquelme, B.; Vergara, M.; Altamirano, C.; Cavieres, M. In vitro Notch-mediated adjuvant immunogenic potency is induced by combining QS-21 and MPL in a co-culture model of PBMC and HUVEC cells. Toxicol. Vitr. 2020, 68, 104947. [Google Scholar] [CrossRef]
- FDA. Coronavirus (COVID-19) Update: FDA Authorizes Emergency Use of Novavax COVID-19 Vaccine, Adjuvanted; FDA: Silver Spring, ML, USA, 2022. [Google Scholar]
- Hauguel, T.M.; Hackett, C.J. Rationally-designed vaccine adjuvants: Separating efficacy from toxicity. Front. Biosci. 2008, 13, 2806–2813. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Batista-Duharte, A.; Portuondo, D.; Carlos, I.Z.; Pérez, O. An approach to local immunotoxicity induced by adjuvanted vaccines. Int. Immunopharmacol. 2013, 17, 526–536. [Google Scholar] [CrossRef]
- Furrie, E.; Smith, R.E.; Turner, M.W.; Strobel, S.; Mowat, A. Induction of local innate immune responses and modulation of antigen uptake as mechanisms underlying the mucosal adjuvant properties of immune stimulating complexes (ISCOMS). Vaccine 2002, 20, 2254–2262. [Google Scholar] [CrossRef]
- Pellegrino, P.; Clementi, E.; Radice, S. On vaccine’s adjuvants and autoimmunity: Current evidence and future perspectives. Autoimmun. Rev. 2015, 14, 880–888. [Google Scholar] [CrossRef]
- Batista-Duharte, A.; Portuondo, D.; Pérez, O.; Carlos, I.Z. Systemic immunotoxicity reactions induced by adjuvanted vaccines. Int. Immunopharmacol. 2014, 20, 170–180. [Google Scholar] [CrossRef]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Zomer, H.D.; Trentin, A.G. Skin wound healing in humans and mice: Challenges in translational research. J. Dermatol. Sci. 2018, 90, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.J.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T.; Nurden, P.; Sanchez, M.; Andia, I.; Anitua, E. Platelets and wound healing. Front. Biosci. 2008, 13, 3532–3548. [Google Scholar] [CrossRef]
- Ley, K. Integration of inflammatory signals by rolling neutrophils. Immunol. Rev. 2002, 186, 8–18. [Google Scholar] [CrossRef]
- Reinke, J.; Sorg, H. Wound Repair and Regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Hübner, G.; Brauchle, M.; Smola, H.; Madlener, M.; Fässler, R.; Werner, S. Differential regulation of pro-inflammatory cytokines during wound healing in normal and glucocorticoid-treated mice. Cytokine 1996, 8, 548–556. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Mark, D.; Banda, M.J.; Werb, Z. Wound Macrophages Express TGF-α and Other Growth Factors in Vivo: Analysis by mRNA Phenotyping. Science 1988, 241, 708–712. [Google Scholar] [CrossRef]
- Velnar, T.; Bailey, T.; Smrkolj, V. The Wound Healing Process: An Overview of the Cellular and Molecular Mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef]
- Gonzalez, A.C.D.O.; Costa, T.F.; de Araújo Andrade, Z.; Medrado, A.R.A.P. Wound Healing—A Literature Review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Kirsner, R. Pathophysiology of acute wound healing. Clin. Dermatol. 2007, 25, 9–18. [Google Scholar] [CrossRef]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in Wound Healing. J. Invest. Dermatol. Symp. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Profyris, C.; Tziotzios, C.; Do Vale, I. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics: Part, I. the molecular basis of scar formation. J. Am. Acad. Dermatol. 2012, 66, 1–10. [Google Scholar] [CrossRef]
- Lazarus, G.S.; Cooper, D.M.; Knighton, D.R.; Margolis, D.J.; Percoraro, R.E.; Rodeheaver, G.; Robson, M.C. Definitions and guidelines for assessment of wounds and evaluation of healing. Arch. Dermatol. 1994, 130, 489–493. [Google Scholar] [CrossRef]
- Gottrup, F. A specialized wound-healing center concept: Importance of a multidisciplinary department structure and surgical treatment facilities in the treatment of chronic wounds. Am. J. Surg. 2004, 187, S38–S43. [Google Scholar] [CrossRef]
- Eming, S.A.; Koch, M.; Krieger, A.; Brachvogel, B.; Kreft, S.; Bruckner-Tuderman, L.; Krieg, T.; Shannon, J.D.; Fox, J.W. Differential Proteomic Analysis Distinguishes Tissue Repair Biomarker Signatures in Wound Exudates Obtained from Normal Healing and Chronic Wounds. J. Proteome Res. 2010, 9, 4758–4766. [Google Scholar] [CrossRef]
- Beidler, S.K.; Douillet, C.D.; Berndt, D.F.; Keagy, B.A.; Rich, P.B.; Marston, W.A. Inflammatory cytokine levels in chronic venous insufficiency ulcer tissue before and after compression therapy. J. Vasc. Surg. 2009, 49, 1013–1020. [Google Scholar] [CrossRef]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef]
- Pulido, T.; Velarde, M.C.; Alimirah, F. The senescence-associated secretory phenotype: Fueling a wound that never heals. Mech. Ageing Dev. 2021, 199, 111561. [Google Scholar] [CrossRef]
- Trengove, N.J.; Stacey, M.C.; Macauley, S.; Bennett, N.; Gibson, J.; Burslem, F.; Murphy, G.; Schultz, G. Analysis of the acute and chronic wound environments: The role of proteases and their inhibitors. Wound Repair Regen. 1999, 7, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.V.; Raffetto, J.D.; Phillips, T.; Menzoian, J.O.; Park, H.-Y. The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: A potential mechanism for senescence in venous ulcers. J. Vasc. Surg. 1999, 30, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, B.; Byun, H.J.; Yu, L.; Nguyen, T.M.; Nguyen, T.H.; Do, P.A.; Kim, E.J.; Cheong, K.A.; Kim, K.S.; et al. Resolvin d1 suppresses H2O2-induced senescence in fibroblasts by inducing autophagy through the mir-1299/arg2/arl1 axis. Antioxidants 2021, 10, 1924. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, F.; Luo, H.; Xu, G.; Wang, D. Inflammatory Microenvironment of Skin Wounds. Front. Immunol. 2022, 13, 789274. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.A.; Sacks, F.; Braunwald, E. Long-Term Effects of Pravastatin on Plasma Concentration of C-reactive Protein. Circulation 1999, 100, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Kaptoge, S.; Seshasai, S.R.K.; Jørgensen, T.; Danesh, J.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef]
- Clearfield, M. C-reactive protein levels and outcomes after statin therapy. Curr. Atheroscler. Rep. 2006, 8, 8–9. [Google Scholar]
- Everett, B.M.; Pradhan, A.D.; Solomon, D.H.; Paynter, N.; MacFadyen, J.; Zaharris, E.; Gupta, M.; Clearfield, M.; Libby, P.; Hasan, A.A.; et al. Rationale and design of the Cardiovascular Inflammation Reduction Trial: A test of the inflammatory hypothesis of atherothrombosis. Am. Heart J. 2013, 166, 199–207.e15. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Fazio, S. From, Lipids to Inflammation: New Approaches to Reducing Atherosclerotic Risk. Circ. Res. 2016, 118, 732–749. [Google Scholar] [CrossRef]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen a phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Testing the inflammatory hypothesis of atherothrombosis: Scientific rationale for the cardiovascular inflammation reduction trial (CIRT). J. Thromb. Haemost. 2009, 7 (Suppl. 1), 332–339. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Spite, M. Specialized pro-resolving mediators in cardiovascular diseases. Mol. Aspects Med. 2017, 58, 65–71. [Google Scholar] [CrossRef]
- Eriksson, E.; Liu, P.Y.; Schultz, G.S.; Martins-Green, M.M.; Tanaka, R.; Weir, D.; Gould, L.J.; Armstrong, D.G.; Gibbons, G.W.; Wolcott, R.; et al. Chronic wounds: Treatment consensus. Wound Repair Regen. 2022, 30, 156–171. [Google Scholar] [CrossRef]
- Frykberg, R.G.; Banks, J. Challenges in the Treatment of Chronic Wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Järbrink, K.; Divakar, U.; Bajpai, R.; Upton, Z.; Schmidtchen, A.; Car, J. The humanistic and economic burden of chronic wounds: A systematic review. Wound Repair Regen. 2019, 27, 114–125. [Google Scholar] [CrossRef]
- Salisbury, A.-M.; Mullin, M.; Foulkes, L.; Chen, R.; Percival, S.L. The Ability of a Concentrated Surfactant Gel to Reduce an Aerobic, Anaerobic and Multispecies Bacterial Biofilm In Vitro. Adv. Exp. Med. Biol. 2021, 1323, 149–157. [Google Scholar]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- FDA. Productos de Terapia Celular y Génica Aprobados; FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 24 July 2022).
- Zhang, Q.-Z.; Su, W.-R.; Shi, S.-H.; Wilder-Smith, P.; Xiang, A.P.; Wong, A.; Nguyen, A.L.; Kwon, C.W.; Le, A.D. Human Gingiva-Derived Mesenchymal Scro-phages and Enhance Cutaneous Wound Healing. Stem Cells 2010, 28, 1856–1868. [Google Scholar] [CrossRef]
- Qi, Y.; Jiang, D.; Sindrilaru, A.; Stegemann, A.; Schatz, S.; Treiber, N.; Rojewski, M.; Schrezenmeier, H.; Vander Beken, S.; Wlaschek, M.; et al. TSG-6 Released from Intradermally Injected Mesenchymal Stem Cells Accelerates Wound Healing and Reduces Tissue Fibrosis in Murine Full-Thickness Skin Wounds. J. Investig. Dermatol. 2014, 134, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Prateeksha, P.; Das, H. Dental Pulp-Derived Stem Cells Reduce Inflammation, Accelerate Wound Healing and Mediate M2 Polarization of Myeloid Cells. Biomedicines 2022, 10, 1999. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2019, 53, e12712. [Google Scholar] [CrossRef]
- Krasilnikova, O.A.; Baranovskii, D.S.; Lyundup, A.V.; Shegay, P.V.; Kaprin, A.D.; Klabukov, I.D. Stem and Somatic Cell Monotherapy for the Treatment of Diabetic Foot Ulcers: Review of Clinical Studies and Mechanisms of Action. Stem Cell Rev. Rep. 2022, 18, 1974–1985. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Han, C.; Sun, X.; Liu, L.; Jiang, H.; Shen, Y.; Xu, X.; Li, J.; Zhang, G.; Huang, J.; Lin, Z.; et al. Exosomes and Their Therapeutic Potentials of Stem Cells. Stem Cells Int. 2016, 2016, 7653489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bai, X.; Zhao, B.; Li, Y.; Zhang, Y.; Li, Z.; Wang, X.; Luo, L.; Han, F.; Zhang, J.; et al. Cell-free therapy based on adipose tissue stem cell-derived exosomes promotes wound healing via the PI3K/Akt signaling pathway. Exp. Cell Res. 2018, 370, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiao, Y.; Pan, Y.; Zhang, L.; Gong, H.; Qi, Y.; Wang, M.; Gong, H.; Shao, M.; Wang, X.; et al. Fetal Dermal Mesenchymal Stem Cell-Derived Exosomes Accelerate Cutaneous Wound Healing by Activating Notch Signaling. Stem Cells Int. 2019, 2019, 2402916–11. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Yang, J.; Yu, Y.; Chai, J.; Wang, L.; Ma, L.; Yin, H. Exosome Derived From Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-induced Excessive Inflammation. EBioMedicine 2016, 8, 72–82. [Google Scholar] [CrossRef]
- Li, X.; Xie, X.; Lian, W.; Shi, R.; Han, S.; Zhang, H.; Lu, L.; Li, M. Exosomes from adipose-derived stem cells overexpressing Nrf2 accelerate cutaneous wound healing by promoting vascularization in a diabetic foot ulcer rat model. Exp. Mol. Med. 2018, 50, 29. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wang, J.; Zhou, X.; Xiong, Z.; Zhao, J.; Yu, R.; Huang, F.; Zhang, H.; Chen, L. Exosomes derived from human adipose mensenchymal stem cells accelerates cutaneous wound healing via optimizing the characteristics of fibroblasts. Sci. Rep. 2016, 6, 32993, Correction on Sci. Rep. 2020, 10, 6693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, J.; Niu, X.; Shangchun, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Fomby, P.; Cherlin, A.J.; Hadjizadeh, A.; Doillon, C.J.; Sueblinvong, V.; Weiss, D.J.; Bates, J.H.; Gilbert, T.; Liles, W.C.; Lutzko, C.; et al. Stem cells and cell therapies in lung biology and diseases: Conference report. Ann. Am. Thorac. Soc. 2010, 12, 181–204. [Google Scholar]
- Pearson, R.G.; Bhandari, R.; Quirk, R.A.; Shakesheff, K.M. Recent Advances in Tissue Engineering. J. Long-Term Eff. Med Implant. 2017, 27, 199–231. [Google Scholar] [CrossRef]
- Metcalfe, A.; Ferguson, M.W.J. Tissue engineering of Mehrabanireplacement skin: The crossroads of biomaterials, wound healing, embryonic development, stem cells and regeneration. J. R. Soc. Interface 2007, 4, 413–437. [Google Scholar] [CrossRef]
- Maksimova, N.V.; Michenko, A.V.; Krasilnikova, O.A.; Klabukov, I.D.; Gadaev, I.Y.; Krasheninnikov, M.E.; Belkov, P.A.; Lyundup, A.V. Mesenchymal stromal cells therapy alone does not lead to the complete restoration of the skin parameters in diabetic foot patients within a 3-year follow-up period. BioImpacts 2022, 12, 51–55. [Google Scholar] [CrossRef]
- Rasouli, M.; Rahimi, A.; Soleimani, M.; Keshel, S.H. The interplay between extracellular matrix and progenitor/stem cells during wound healing: Opportunities and future directions. Acta Histochem. 2021, 123, 151785. [Google Scholar] [CrossRef]
- Powers, J.G.; Morton, L.M.; Phillips, T.J. Dressings for chronic wounds. Dermatol. Ther. 2013, 26, 197–206. [Google Scholar] [CrossRef]
- Weinstein-Oppenheimer, C.R.; Aceituno, A.R.; Brown, D.I.; Acevedo, C.; Ceriani, R.; Fuentes, M.A.; Albornoz, F.; Henriquez-Roldán, C.F.; Morales, P.; Maclean, C.; et al. The effect of an autologous cellular gel-matrix integrated implant system on wound healing. J. Transl. Med. 2010, 8, 59. [Google Scholar] [CrossRef]
- Weinstein-Oppenheimer, C.R.; Brown, D.I.; Coloma, R.; Morales, P.; Reyna-Jeldes, M.A.; Díaz, M.J.; Sánchez, E.; Acevedo, C.A. Design of a hybrid biomaterial for tissue engineering: Biopolymer-scaffold integrated with an autologous hydrogel carrying mesenchymal stem-cells. Mater. Sci. Eng. C 2017, 79, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.L.; Huang, H.H.; Chen, T.; Ju, K.C.; Kuo, S.M. Angiogenesis-promoting effect of LIPUS on hADSCs and HUVECs cultured on collagen/hyaluronan scaffolds. Mater. Sci. Eng. C 2019, 102, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, S.S.; Mohammadi, A.A.; Kabiri, H.; Hashempoor, M.R.; Mahmoodi, M.; Amini, M.; Mehrabani, D. The healing effect of Wharton’s jelly stem cells seeded on biological scaffold in chronic skin ulcers: A randomized clinical trial. J. Cosmet. Dermatol. 2019, 18, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhong, Y.; Hu, H.; Shao, D.; Haag, R.; Schirner, M.; Lee, J.; Sullenger, B.; Leong, K.W. Design of therapeutic biomaterials to control inflammation. Nat. Rev. Mater. 2022, 7, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, A.; Dey, A.D.; Behl, T.; Chadha, S. Stem cells and growth factors-based delivery approaches for chronic wound repair and regeneration: A promise to heal from within. Life Sci. 2021, 268, 118932. [Google Scholar] [CrossRef]
- Hazrati, R.; Davaran, S.; Omidi, Y. Bioactive functional scaffolds for stem cells delivery in wound healing and skin regeneration. React. Funct. Polym. 2022, 174, 105233. [Google Scholar] [CrossRef]
- Zeng, X.; Chen, B.; Wang, L.; Sun, Y.; Jin, Z.; Liu, X.; Ouyang, L.; Liao, Y. Chitosan Puerarin hydrogel for accelerated wound healing in diabetic subjects by miR-29ab1 mediated inflammatory axis suppression. Bioact. Mater. 2023, 19, 653–665. [Google Scholar] [CrossRef]
- Ravishankar, K.; Venkatesan, M.; Desingh, R.P.; Mahalingam, A.; Sadhasivam, B.; Subramaniyam, R.; Dhamodharan, R. Biocompatible hydrogels of chitosan-alkali lignin for potential wound healing applications. Mater. Sci. Eng. C 2019, 102, 447–457. [Google Scholar] [CrossRef]
- Turner, P.R.; McConnell, M.; Young, S.L.; Cabral, J.D. 3D living dressing improves healing and modulates immune response in a thermal injury model. Tissue Eng. Part C: Methods 2022, 28, 431–439. [Google Scholar] [CrossRef]
- Heras, K.L.; Igartua, M.; Santos-Vizcaino, E.; Hernandez, R.M. Cell-based dressings: A journey through chronic wound management. Biomater. Adv. 2022, 135, 212738. [Google Scholar] [CrossRef]
- Geesala, R.; Bar, N.; Dhoke, N.R.; Basak, P.; Das, A. Data on bone marrow stem cells delivery using porous polymer scaffold. Data Brief 2015, 6, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Pujana, A.; Vining, K.H.; Zhang, D.K.; Santos-Vizcaino, E.; Igartua, M.; Hernandez, R.M.; Mooney, D.J. Multifunctional biomimetic hydrogel systems to boost the immunomodulatory potential of mesenchymal stromal cells. Biomaterials 2020, 257, 120266. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, H.; Ma, Z.; Qiang, L.; Yang, Z.; Yang, X.; Zhou, X.; Dai, K.; Wang, J. Mussel-Inspired Nanostructures Potentiate the Immunomodulatory Properties and Angiogenesis of Mesenchymal Stem Cells. ACS Appl. Mater. Interfaces 2019, 11, 17134–17146. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Mao, J.; Yao, K.; Yang, G.; Cui, L.; Cao, Y. A study on a chitosan-gelatin-hyaluronic acid scaffold as artificial skin in vitro and its tissue engineering applications. J. Biomater. Sci. Polym. Ed. 2004, 15, 25–40. [Google Scholar] [CrossRef]
- Serhan, C.N. Systems approach to inflammation resolution: Identification of novel anti-inflammatory and pro-resolving mediators. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 44–48. [Google Scholar] [CrossRef]
- Serhan, C.N. Resolution Phase of Inflammation: Novel Endogenous Anti-Inflammatory and Proresolving Lipid Mediators and Pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef]
- Serhan, C.N. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol. Aspects Med. 2017, 58, 1–11. [Google Scholar] [CrossRef]
- Miyahara, T.; Runge, S.; Chatterjee, A.; Chen, M.; Mottola, G.; Fitzgerald, J.M.; Serhan, C.N.; Conte, M.S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013, 27, 2220–2232. [Google Scholar] [CrossRef]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and anti-inflammatory actions: Pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef]
- Menon, R.; Krzyszczyk, P.; Berthiaume, F. Pro-Resolution Potency of Resolvins D1, D2 and E1 on Neutrophil Migration and in Dermal Wound Healing. Nano Life 2017, 7, 1750002. [Google Scholar] [CrossRef]
- Artiach, G.; Carracedo, M.; Clària, J.; Laguna-Fernandez, A.; Bäck, M. Opposing Effects on Vascular Smooth Muscle Cell Proliferation and Macrophage-induced Inflammation Reveal a Protective Role for the Proresolving Lipid Mediator Receptor ChemR23 in Intimal Hyperplasia. Front. Pharmacol. 2018, 9, 1327. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Aspects Med. 2017, 58, 114–129. [Google Scholar] [CrossRef]
- Chiang, N.; Libreros, S.; Norris, P.C.; De La Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immu-noresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, A.F.; Claudino, R.F.; Dutra, R.C.; Marcon, R.; Calixto, J.B. Omega-3 Fatty Acid-Derived Mediators 17(R)-Hydroxy Docosahexaenoic Acid, Aspirin-Triggered Resolvin D1 and Resolvin D2 Prevent Experimental Colitis in Mice. J. Immunol. 2011, 187, 1957–1969. [Google Scholar] [CrossRef]
- Kurihara, T.; Jones, C.N.; Yu, Y.-M.; Fischman, A.J.; Watada, S.; Tompkins, R.G.; Fagan, S.P.; Irimia, D. Resolvin D2 restores neutrophil directionality and improves survival after burns. FASEB J. 2013, 27, 2270–2281. [Google Scholar] [CrossRef]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Sharma, A.; Chen, M.; Toy, R.; Mottola, G.; Conte, M.S. The Pro-Resolving Lipid Mediator Maresin 1 (MaR1) Attenuates Inflammatory Signaling Pathways in Vascular Smooth Muscle and Endothelial Cells. PLoS ONE 2014, 9, e113480. [Google Scholar]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Soehnlein, O. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef]
- Kantarci, A.; Aytan, N.; Palaska, I.; Stephens, D.; Crabtree, L.; Benincasa, C.; Jenkins, B.G.; Carreras, I.; Dedeoglu, A. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Exp. Neurol. 2018, 300, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Cantú, S.M.; Lee, H.J.; Donoso, A.; Puyó, A.M.; Peredo, H.A. El Ácido Araquidónico Y Sus Derivados. Generalidades De Los Prostanoides En Relación Con Procesos Inflamatorios. Cienc. Investig. 2017, 67, 5–12. [Google Scholar]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Wali, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar] [PubMed]
- Li, Q.F.; Hao, H.; Tu, W.S.; Guo, N.; Zhou, X.Y. Maresins: Anti-inflammatory pro-resolving mediators with therapeutic potential. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7442–7453. [Google Scholar] [PubMed]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2015, 1851, 397–413. [Google Scholar] [CrossRef]
- Andrews, D.; Godson, C. Lipoxins and synthetic lipoxin mimetics: Therapeutic potential in renal diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158940. [Google Scholar] [CrossRef]
- Liu, G.; Liu, Q.; Shen, Y.; Kong, D.; Gong, Y.; Tao, B.; Chen, G.; Guo, S.; Li, J.; Zuo, S.; et al. Early treatment with Resolvin E1 facilitates myocardial recovery from ischaemia in mice. Br. J. Pharmacol. 2018, 175, 1205–1216. [Google Scholar] [CrossRef]
- Sato, M.; Aoki-Saito, H.; Fukuda, H.; Ikeda, H.; Koga, Y.; Yatomi, M.; Tsurumaki, H.; Maeno, T.; Saito, T.; Nakakura, T.; et al. Resolvin E3 attenuates allergic airway inflammation via the interleukin-23-interleukin-17A pathway. FASEB J. 2019, 33, 12750–12759. [Google Scholar] [CrossRef]
- Makino, Y.; Miyahara, T.; Nitta, J.; Miyahara, K.; Seo, A.; Kimura, M.; Suhara, M.; Akai, A.; Akagi, D.; Yamamoto, K.; et al. Proresolving lipid mediators resolvin D1 and protectin D1 isomer attenuate neointimal hyperplasia in the rat carotid artery balloon injury model. J. Surg. Res. 2019, 233, 104–110. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Bin Deng, B.; Wang, C.-W.; Arnardottir, H.H.; Li, Y.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N. Maresin Biosynthesis and Identification of Maresin 2, a New Anti-Inflammatory and Pro-Resolving Mediator from Human Macrophages. PLoS ONE 2014, 9, e102362. [Google Scholar]
- Abbott, A. Cell culture: Biology’s new dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Cell Therapy for Diabetic Foot Ulcer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05234086 (accessed on 7 July 2022).
- Serhan, C.N.; Levy, B.D. Resolvinas en Inflamación: Emergencia de la superfamilia de mediadores de resolución pro. Investig. Clínic. 2018, 128, 2657–2669. [Google Scholar]
- Winkler, J.; Libreros, S.; De La Rosa, X.; Sansbury, B.; Norris, P.; Chiang, N.; Fichtner, D.; Keyes, G.S.; Wourms, N.; Spite, M.; et al. Structural insights into Resolvin D4 actions and further metabolites via a new total organic synthesis and validation. J. Leukoc. Biol. 2018, 103, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Potent Immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and Protectins in Inflammation Resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef]
- Orr, S.K.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N. Proresolving actions of a new resolvin D1 analog mimetic qualifies as an immu-noresolvent. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L904–L911. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cells | Phase | Sponsor | Description |
|---|---|---|---|
| Stromal fraction | I | Antria | Stromal Vascular Fraction from Lipoaspirate in the Treatment of Chronic Non-healing Wounds |
| SkinTE: keratinocytes, dermal fibroblasts, dermal endothelial cells, and follicular cells, as well as extracellular matrix. | III | PolarityTE | Multi-Center, Prospective, Randomized Controlled Trial Evaluating SkinTE® in the Treatment of Wagner 2 Diabetic Food Ulcer |
| ABCB5-positive dermal mesenchymal stromal cells | IIB | RHEACELL GmbH & Co. KG | Allogeneic ABCB5-positive Dermal Mesenchymal Stromal Cells for Treatment of Chronic Venous Ulcers |
| Mesenchymal stem cells | I | University of Colorado, Denver | Open-Label Safety Study of Umbilical Cord Lining Mesenchymal Stem Cells (Corlicyte®) To Heal Chronic Diabetic Foot Ulcers |
| Mesenchymal stem cells | II | Instituto para el Desarrollo Biotecnológico y la Innovación | Cell Therapy for Diabetic Foot Ulcer |
| Umbilical cord blood mononuclear cells | III | Peking University Third Hospital | Efficacy and Safety of Umbilical Cord Blood Mononuclear Cell Gel in the Treatment of Refractory Diabetic Foot Ulcer. |
| Mesenchymal stem cells | I | Beijing Tongren Hospital | Human Placental Mesenchymal Stem Cells Treatment on Diabetic Foot Ulcer |
| Mesenchymal stem cells | II | Anterogen Co., Ltd. | Efficacy and Safety of ALLO-ASC-SHEET in Subjects with Diabetic Foot Ulcers. It is a hydrogel with allogenic adipocytes-derived mesenchymal cells |
| Ligands | Receptors | Target cells | Function | References |
|---|---|---|---|---|
| RvE1 | ChemR23 BLT1 | Monocytes, macrophages, dendritic cells and myeloid cells. | ↑ phagocytosis by macrophages. Limits neutrophil signals. ↑ PMN apoptosis. ↓ IL-12 production. It inhibits the release of IL-23. ↑ expression of CCR5 that contributes to the mobility of leukocytes including T cells. | [165] |
| RvE2 | ChemR23 BLT1 | Monocytes, macrophages, dendritic cells and myeloid cells. | ↓ neutrophil chemotaxis. ↑ phagocytosis of apoptotic neutrophils. ↑ phagocytosis of bacteria. ↑ efferocytosis. | [146] |
| RvE3 | --- | Neutrophils | Stops neutrophil chemotaxis. ↓ IL-4, IL-5, IL-13, IL-17, and IL-23 mRNA levels in lung cell culture. | [146] |
| RvD1 | GPR32 (↑ affinity) ALX/FPR2 | Leukocyte and macrophages. | ↑ phagocytosis of macrophages and apoptotic leukocytes. ↑ efferocytosis. ↑ polarization of M2 macrophages. | [157] |
| RvD2 | GRP18 | Monocytes, macrophages and neutrophils. | ↑ phagocytosis of apoptotic neutrophils. ↓ movement of neutrophils ↑ efferocytosis. Protective role against sepsis, since it favors phagocytosis of bacteria. | [166] |
| RvD3 | ALX/FPR2 GPR32 | Monocytes, macrophages and neutrophils. | ↑ phagocytosis of apoptotic cells ↓ migration of neutrophils. ↑ efferocytosis. ↓ release of IL-6 and IL-23. ↑ IL-10 levels. | [164] |
| RvD4 | --- | Neutrophils. | ↓ neutrophil infiltration. ↑ efferocytosis. It inhibits the production of cytokines. Regulates diapedesis of leukocytes. | [159] |
| RvD5 | GPR32 | Macrophages. | ↑ phagocytosis by macrophages. | [166] |
| LXA4 | ALX/FPR2 GPR32 | Monocytes, macrophages, epithelia, endothelia, and fibroblasts. | They limit neutrophil recruitment and PMN activation. They promote the chemotaxis of monocytes and their infiltration in infectious foci. ↑ efferocytosis. ↑ phenotypic shift of macrophages towards M2. ↑ release of IL-10 and TGF-β. | [167] |
| LXB4 | --- | --- | Biological activity very similar to that of LXA4. | [167] |
| MaR1 | LGR6 BLT1 (↓ affinity) | Macrophages, monocytes, and PMNs. | ↑ phagocytosis ↑ efferocytosis Regulates PMN chemotaxis. ↓ neuropathic pain. ↑ tissue regeneration. ↓ expression of IL-5 and IL-13. ↓ proliferation, migration and differentiation of fibroblasts.It stimulates phenotypic change of macrophages towards M2. | [147,168] |
| MaR2 | --- | Macrophages, monocytes, and PMNs. | ↑ phagocytosis. ↓ PMN infiltration. | [162,166] |
| PD1 | --- | Leukocytes, macrophages and neutrophils. | Inhibits infiltration of polymorphonuclear leukocytes. ↓ expression of TNF-α and IL-6. ↑IL-10 release. | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfaro, S.; Acuña, V.; Ceriani, R.; Cavieres, M.F.; Weinstein-Oppenheimer, C.R.; Campos-Estrada, C. Involvement of Inflammation and Its Resolution in Disease and Therapeutics. Int. J. Mol. Sci. 2022, 23, 10719. https://doi.org/10.3390/ijms231810719
Alfaro S, Acuña V, Ceriani R, Cavieres MF, Weinstein-Oppenheimer CR, Campos-Estrada C. Involvement of Inflammation and Its Resolution in Disease and Therapeutics. International Journal of Molecular Sciences. 2022; 23(18):10719. https://doi.org/10.3390/ijms231810719
Chicago/Turabian StyleAlfaro, Sebastián, Vania Acuña, Ricardo Ceriani, María Fernanda Cavieres, Caroline Ruth Weinstein-Oppenheimer, and Carolina Campos-Estrada. 2022. "Involvement of Inflammation and Its Resolution in Disease and Therapeutics" International Journal of Molecular Sciences 23, no. 18: 10719. https://doi.org/10.3390/ijms231810719