
Interaction of miR-155 with Human Serum Albumin: An Atomic Force Spectroscopy, Fluorescence, FRET, and Computational Modelling Evidence

Abstract
:1. Introduction
2. Results and Discussion
2.1. AFS Results
2.2. Static and Time-Resolved Fluorescence Results
2.3. FRET Results
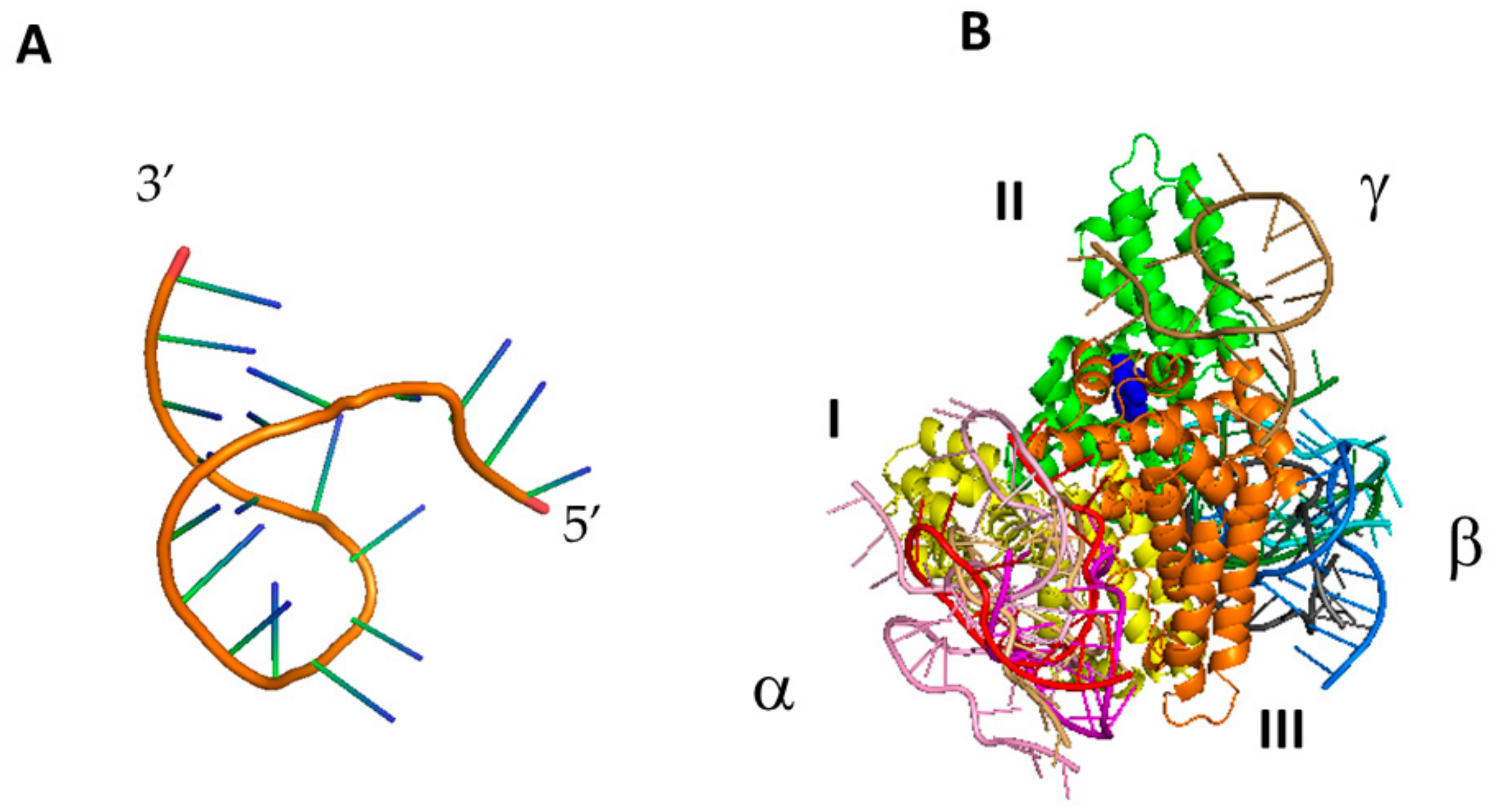
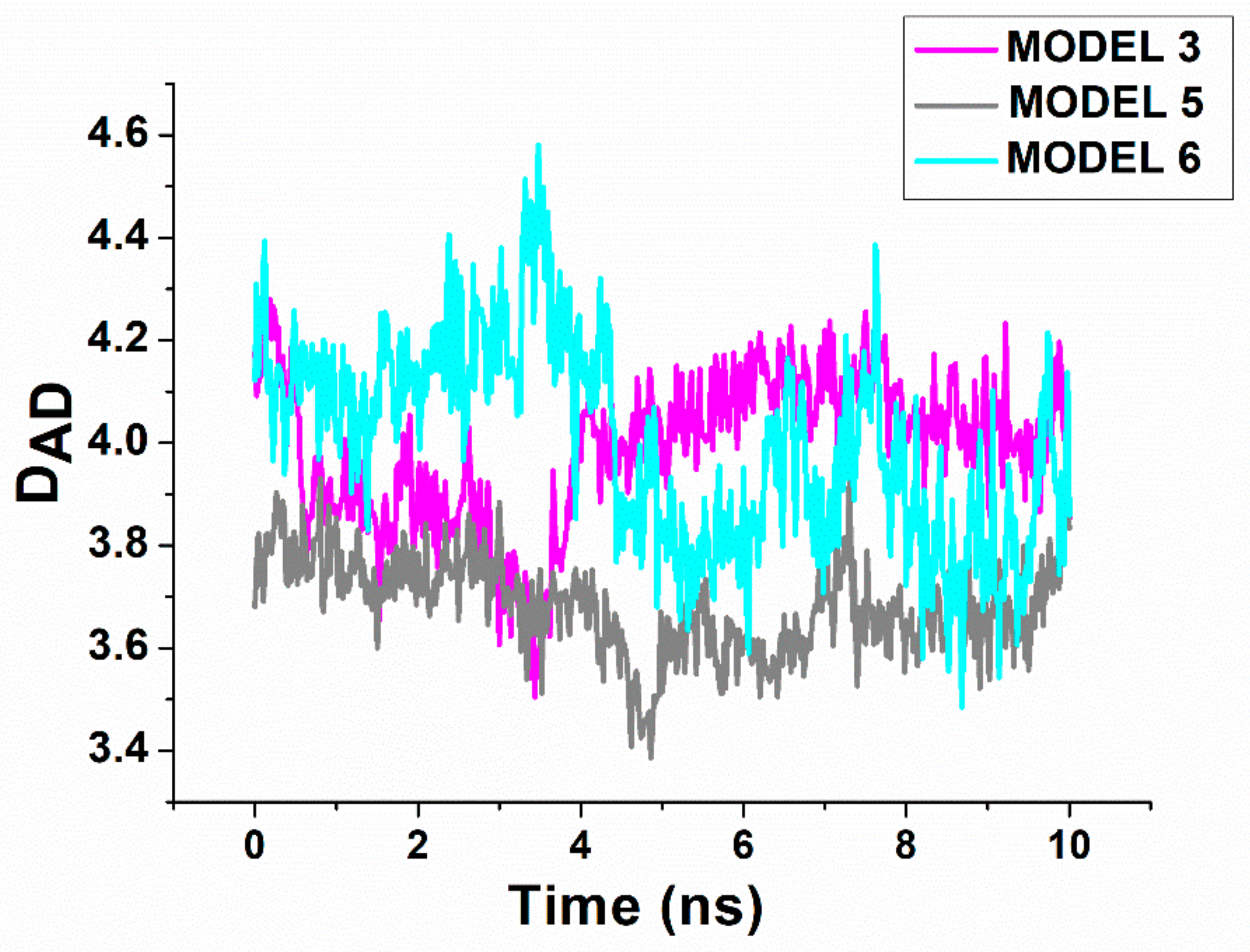
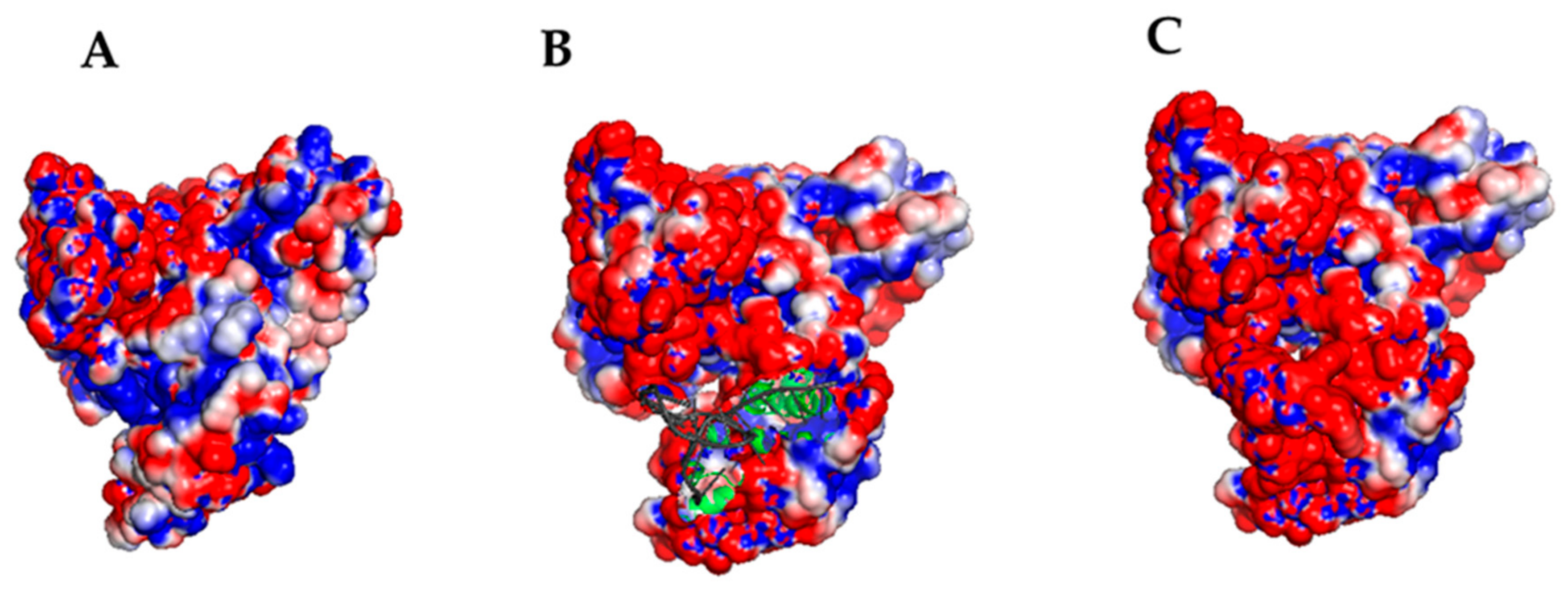
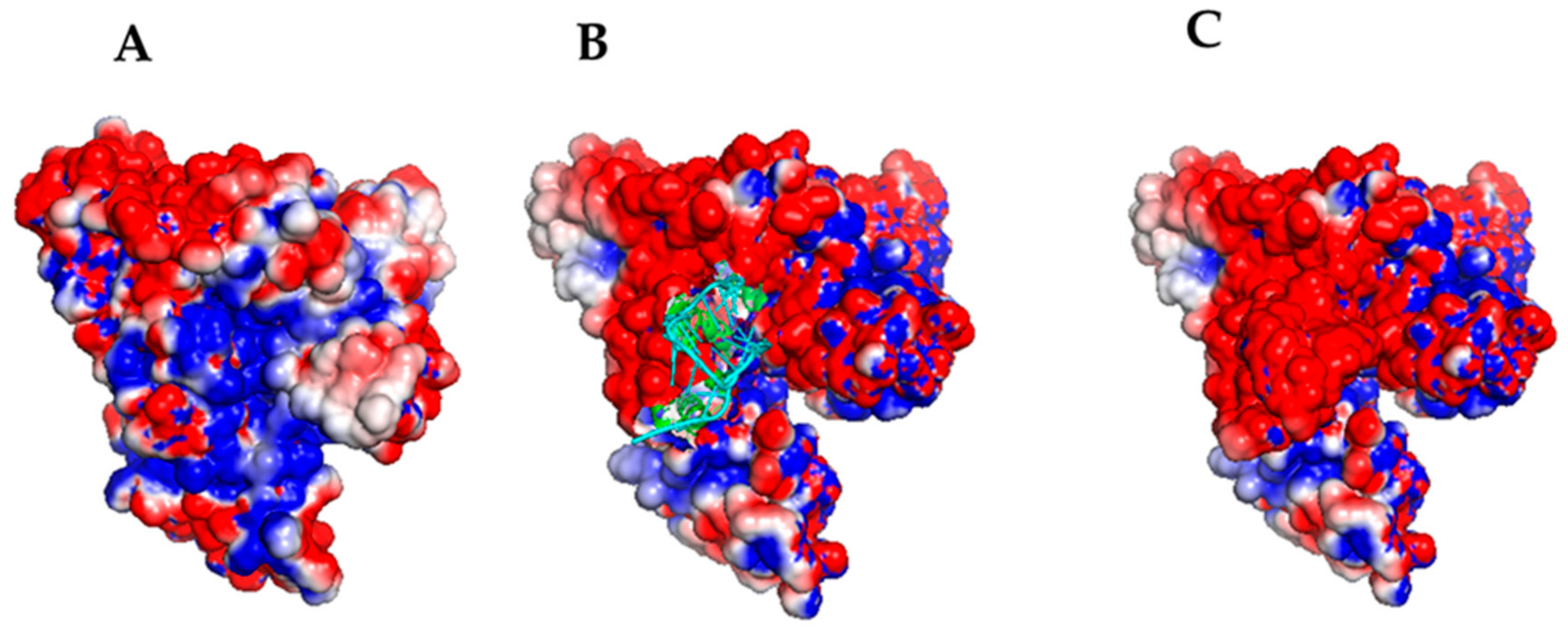
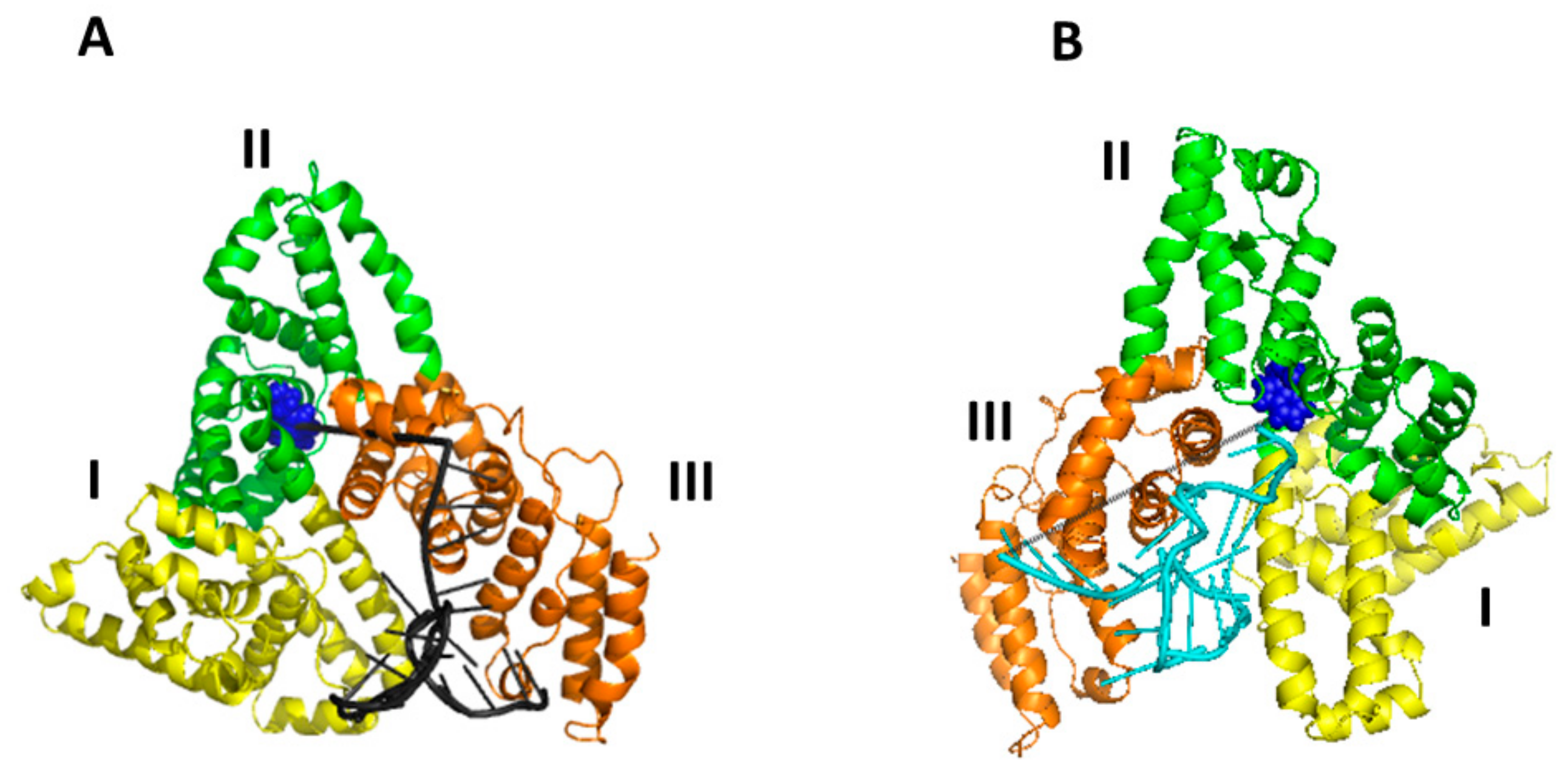
2.4. Computational Docking
3. Materials and Methods
3.1. Materials
3.2. AFS Experiments
3.3. Fluorescence
3.3.1. Static Fluorescence
3.3.2. Time-Resolved Fluorescence
3.3.3. FRET
3.4. Modelling Procedures
3.5. Molecular Dynamics (MD) Simulations
3.6. Calculation of the Binding Free Energy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta-Mol. Cell Res. 2010, 1803, 1231–1243. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. The Two Faces of miRNA. Science 2007, 318, 1877–1878. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef]
- Weiss, C.N.; Ito, K. Chapter Three—A Macro View of MicroRNAs: The Discovery of MicroRNAs and Their Role in Hematopoiesis and Hematologic Disease. In MiRNAs in Aging and Cancer; Galluzzi, L., Vitale, I., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 334, pp. 99–175. ISBN 1937-6448. [Google Scholar]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Shah, V.; Shah, J. Recent trends in targeting miRNAs for cancer therapy. J. Pharm. Pharmacol. 2020, 72, 1732–1749. [Google Scholar] [CrossRef]
- Moscetti, I.; Cannistraro, S.; Bizzarri, A.R. Probing direct interaction of oncomiR-21-3p with the tumor suppressor p53 by fluorescence, FRET and atomic force spectroscopy. Arch. Biochem. Biophys. 2019, 671, 35–41. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Cannistraro, S. Investigation of a Direct Interaction between miR4749 and the Tumor Suppressor p53 by Fluorescence, FRET and Molecular Modeling. Biomolecules 2020, 10, 346. [Google Scholar] [CrossRef]
- Li, J.; Aung, L.H.H.; Long, B.; Qin, D.; An, S.; Li, P. miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci. Rep. 2015, 5, 16422. [Google Scholar] [CrossRef]
- Mishra, V.; Heath, R.J. Structural and Biochemical Features of Human Serum Albumin Essential for Eukaryotic Cell Culture. Int. J. Mol. Sci. 2021, 22, 8411. [Google Scholar] [CrossRef]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef]
- Parodi, A.; Miao, J.; Soond, S.M.; Rudzińska, M.; Zamyatnin, A.A. Albumin Nanovectors in Cancer Therapy and Imaging. Biomolecules 2019, 9, 218. [Google Scholar] [CrossRef]
- Hu, Y.-J.; Liu, Y.; Sun, T.-Q.; Bai, A.-M.; Lü, J.-Q.; Pi, Z.-B. Binding of anti-inflammatory drug cromolyn sodium to bovine serum albumin. Int. J. Biol. Macromol. 2006, 39, 280–285. [Google Scholar] [CrossRef]
- Seedher, N.; Bhatia, S. Reversible binding of celecoxib and valdecoxib with human serum albumin using fluorescence spectroscopic technique. Pharmacol. Res. 2006, 54, 77–84. [Google Scholar] [CrossRef]
- Ha, E.; Bang, J.-H.; Son, J.N.; Cho, H.-C.; Mun, K.-C. Carbamylated albumin stimulates microRNA-146, which is increased in human renal cell carcinoma. Mol. Med. Rep. 2010, 3, 275–279. [Google Scholar]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 497–505. [Google Scholar] [CrossRef]
- Mahesh, G.; Biswas, R. MicroRNA-155: A Master Regulator of Inflammation. J. Interf. Cytokine Res. 2019, 39, 321–330. [Google Scholar] [CrossRef]
- Botti, V.; Marrone, S.; Cannistraro, S.; Bizzarri, A.R. Interaction between miR4749 and Human Serum Albumin as Revealed by Fluorescence, FRET, Atomic Force Spectroscopy and Computational Modelling. Int. J. Mol. Sci. 2022, 23, 1291. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Cannistraro, S. The application of atomic force spectroscopy to the study of biological complexes undergoing a biorecognition process. Chem. Soc. Rev. 2010, 39, 734–749. [Google Scholar] [CrossRef]
- Allison, D.P.; Mortensen, N.P.; Sullivan, C.J.; Doktycz, M.J. Atomic force microscopy of biological samples. Rev. Nanomed. Nanobiotechnol. 2010, 2, 618–634. [Google Scholar] [CrossRef]
- Morfill, J.; Blank, K.; Zahnd, C.; Luginbühl, B.; Kühner, F.; Gottschalk, K.E.; Plückthun, A.; Gaub, H.E. Affinity-matured recombinant antibody fragments analyzed by single-molecule force spectroscopy. Biophys. J. 2007, 93, 3583–3590. [Google Scholar] [CrossRef]
- Bell, G.I. Models for the specific adhesion of cells to cells. Science 1978, 200, 618–627. [Google Scholar] [CrossRef]
- Evans, E. Probing the Relation Between Force—Lifetime—and Chemistry in Single Molecular Bonds. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 105. [Google Scholar] [CrossRef]
- Bonanni, B.; Kamruzzahan, A.S.M.; Bizzarri, A.R.; Rankl, C.; Gruber, H.J.; Hinterdorfer, P.; Cannistraro, S. Single Molecule Recognition between Cytochrome C 551 and Gold-Immobilized Azurin by Force Spectroscopy. Biophys. J. 2005, 89, 2783–2791. [Google Scholar] [CrossRef]
- Funari, G.; Domenici, F.; Nardinocchi, L.; Puca, R.; D’Orazi, G.; Bizzarri, A.R.; Cannistraro, S. Interaction of p53 with Mdm2 and azurin as studied by atomic force spectroscopy. J. Mol. Recognit. 2010, 23, 343–351. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Probing the interaction between p53 and the bacterial protein azurin by single molecule force spectroscopy. J. Mol. Recognit. 2008, 21, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Moscetti, I.; Teveroni, E.; Moretti, F.; Bizzarri, A.R.; Cannistraro, S. MDM2–MDM4 molecular interaction investigated by atomic force spectroscopy and surface plasmon resonance. Int. J. Nanomed. 2016, 11, 4221–4229. [Google Scholar]
- Teale, F.W.J.; Weber, G. Ultraviolet fluorescence of the aromatic amino acids. Biochem. J. 1957, 65, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Centre, N.; Centre, N. Revisitation of FRET methods to measure intraprotein distances in Human Serum Albumin. J. Lumin. 2016, 179, 322–327. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Bhati, A.P.; Zasada, S.J.; Coveney, P.V. Rapid, accurate, precise and reproducible ligand-protein binding free energy prediction. Interface Focus 2020, 10, 20200007. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Lin, T.H. A molecular dynamics study on binding recognition between several 4,5 and 4,6-linked aminoglycosides with A-site RNA. J. Mol. Recognit. 2010, 23, 423–434. [Google Scholar] [CrossRef]
- Ganser, L.R.; Kelly, M.L.; Herschlag, D.; Al-Hashimi, H.M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 2019, 20, 474–489. [Google Scholar] [CrossRef]
- Wu, Y.; Cao, Z.; Yi, H.; Jiang, D.; Mao, X.; Liu, H.; Li, W. Simulation of the interaction between ScyTx and small conductance calcium-activated potassium channel by docking and MM-PBSA. Biophys. J. 2004, 87, 105–112. [Google Scholar] [CrossRef]
- Fuxreiter, M. Electrostatics tunes protein interactions to context. Proc. Natl. Acad. Sci. USA 2022, 119, e2209201119. [Google Scholar] [CrossRef]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868. [Google Scholar] [CrossRef]
- Friedsam, C.; Wehle, A.K.; Kühner, F.; Gaub, H.E. Dynamic single-molecule force spectroscopy: Bond rupture analysis with variable spacer length. J. Phys. Condens. Matter 2003, 15, S1709–S1723. [Google Scholar] [CrossRef]
- Hinterdorfer, P.; Baumgartner, W.; Gruber, H.J.; Schilcher, K.; Schindler, H. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 3477–3481. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Cannistraro, S. 1/Fα Noise in the Dynamic Force Spectroscopy Curves Signals the Occurrence of Biorecognition. Phys. Rev. Lett. 2013, 110, 048104. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; AlSadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. Elife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Zauner, G.; Lonardi, E.; Bubacco, L.; Aartsma, T.J. Tryptophan-to-Dye Fluorescence Energy Transfer Applied to Oxygen Sensing by Using Type-3 Copper Proteins. Chem.–Eur. J. 2007, 13, 7085–7090. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112–e112. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Humphrey, W.F.; Dalke, A.; Schulten, K. VMD—visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force Fields for Protein Simulations. In Protein Simulations; Academic Press: Cambridge, MA, USA, 2003; Volume 66, pp. 27–85. ISBN 0065-3233. [Google Scholar]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Chem. Phys. 1981, 91, 6269–6271. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein-Protein and Protein-DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kholmurodov, K.; Smith, W.; Yasuoka, K.; Darden, T.; Ebisuzaki, T. A smooth-particle mesh Ewald method for DL_POLY molecular dynamics simulation package on the Fujitsu VPP700. J. Comput. Chem. 2000, 21, 1187–1191. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Moscetti, I.; Cannistraro, S. BBA-General Subjects Interaction of the anticancer p28 peptide with p53-DBD as studied by fl uorescence, FRET, docking and MD simulations. BBA-Gen. Subj. 2019, 1863, 342–350. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Basdevant, N.; Weinstein, H.; Ceruso, M. Thermodynamic Basis for Promiscuity and Selectivity in Protein-Protein Interactions: PDZ Domains, a Case Study. J. Am. Chem. Soc. 2006, 128, 12766–12777. [Google Scholar] [CrossRef]
- Ganoth, A.; Friedman, R.; Nachliel, E.; Gutman, M. A molecular dynamics study and free energy analysis of complexes between the Mlc1p protein and two IQ motif peptides. Biophys. J. 2006, 91, 2436–2450. [Google Scholar] [CrossRef]
- Chong, L.T.; Duan, Y.; Wang, L.; Massova, I.; Kollman, P.A. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48G7. Proc. Natl. Acad. Sci. USA 1999, 96, 14330–14335. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MODEL # | DDA in (nm) | DDA ave (nm) | ΔGnonpol solv (kJ/mol) | ΔEMM (kJ/mol) | −TΔS (kJ/mol) | ΔGpol solv (kJ/mol) | ΔGB (kJ/mol) |
|---|---|---|---|---|---|---|---|
| Model 3 | 3.8 | 3.8 ±0.1 | −31 | −175 | 872 | −1300 | −634 |
| Model 5 | 4.0 | 3.7 ± 0.1 | −30 | −198 | 878 | −1600 | −950 |
| Model 6 | 4.2 | 4.0 | −10 | −392 | 919 | −2400 | −1883 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botti, V.; Cannistraro, S.; Bizzarri, A.R. Interaction of miR-155 with Human Serum Albumin: An Atomic Force Spectroscopy, Fluorescence, FRET, and Computational Modelling Evidence. Int. J. Mol. Sci. 2022, 23, 10728. https://doi.org/10.3390/ijms231810728
Botti V, Cannistraro S, Bizzarri AR. Interaction of miR-155 with Human Serum Albumin: An Atomic Force Spectroscopy, Fluorescence, FRET, and Computational Modelling Evidence. International Journal of Molecular Sciences. 2022; 23(18):10728. https://doi.org/10.3390/ijms231810728
Chicago/Turabian StyleBotti, Valentina, Salvatore Cannistraro, and Anna Rita Bizzarri. 2022. "Interaction of miR-155 with Human Serum Albumin: An Atomic Force Spectroscopy, Fluorescence, FRET, and Computational Modelling Evidence" International Journal of Molecular Sciences 23, no. 18: 10728. https://doi.org/10.3390/ijms231810728