

Inhibition of FGFR Signaling by Targeting FGF/FGFR Extracellular Interactions: Towards the Comprehension of the Molecular Mechanism through NMR Approaches

 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
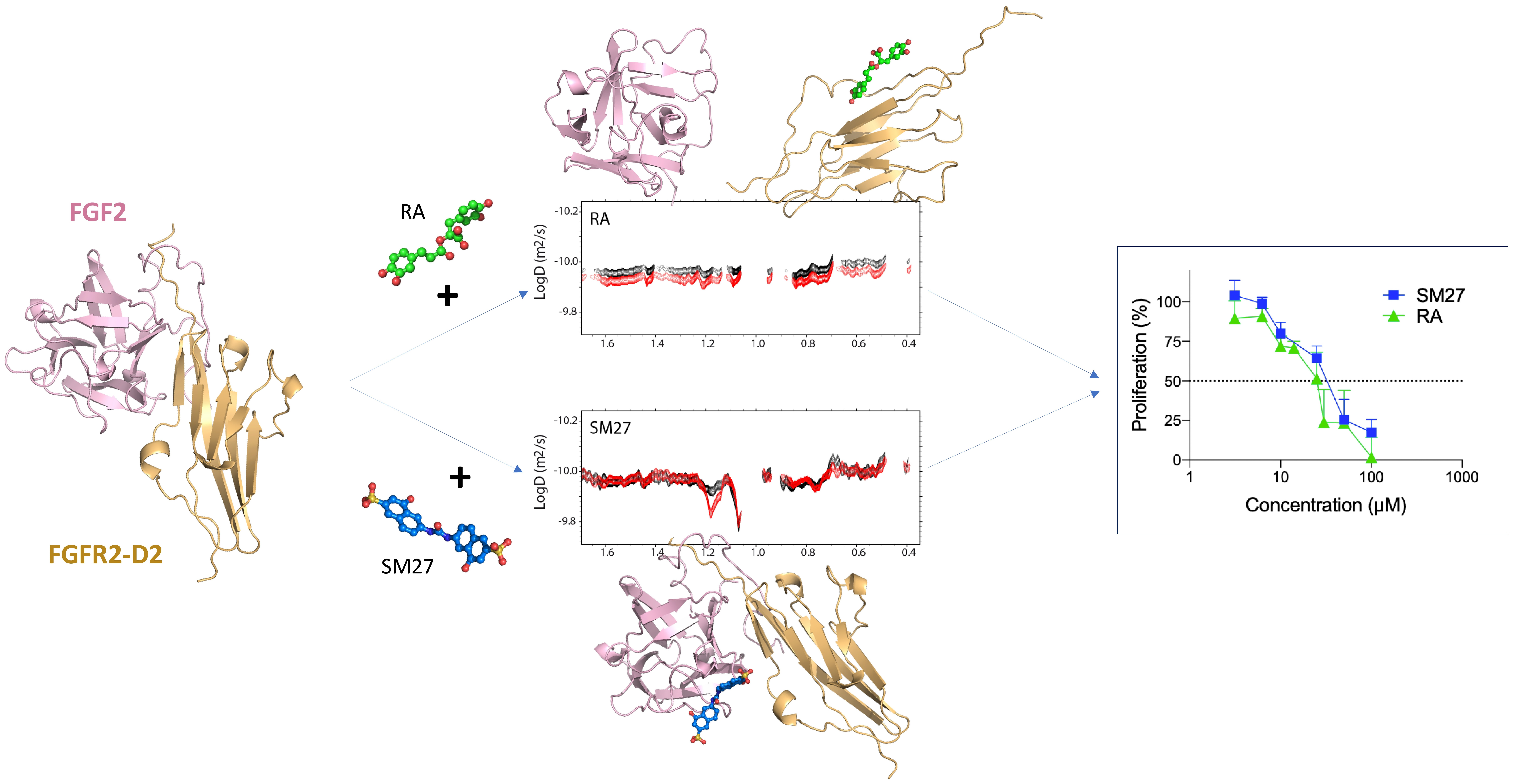
2.1. RA, RES, SM27, and DOBE Establish Direct Interactions with the Extracellular FGF2/D2 Domain
2.2. RA, RES, SM27 Inhibit FGF2-Induced Endothelial Cell Proliferation
2.3. A Diffusion-Ordered NMR Spectroscopy Analysis Highlights the Efficacy of Ligands upon Complex Dissociation
2.4. Preferential Binding of Ligands to FGF2 or D2
2.4.1. A Ligand-Based Point of View
2.4.2. A Protein-Based Point of View
3. Materials and Methods
3.1. Samples
3.2. NMR Experiments
3.3. Endothelial Cell Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chioni, A.-M.; Grose, R.P. Biological Significance and Targeting of the FGFR Axis in Cancer. Cancers 2021, 13, 5681. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers 2021, 13, 5796. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2018, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2020, 124, 880–892. [Google Scholar] [CrossRef]
- Yang, T.; Liang, L.; Wang, M.-D.; Shen, F. FGFR inhibitors for advanced cholangiocarcinoma. Lancet Oncol. 2020, 21, 610–612. [Google Scholar] [CrossRef]
- Katoh, M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef]
- Mohammadi, M.; McMahon, G.; Sun, L.; Tang, C.; Hirth, P.; Yeh, B.K.; Hubbard, S.R.; Schlessinger, J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural basis for FGF receptor dimerization and activation. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Salituro, F.G.; Blanco, M.-J. Impact of Allosteric Modulation in Drug Discovery: Innovation in Emerging Chemical Modalities. ACS Med. Chem. Lett. 2020, 11, 1810–1819. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Mitchell, K.; Siebzehnrubl, F.A.; Lathia, J.D. FGF2: A novel druggable target for glioblastoma? Expert Opin. Ther. Targets 2020, 24, 311–318. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular Mechanism of SSR128129E, an Extracellularly Acting, Small-Molecule, Allosteric Inhibitor of FGF Receptor Signaling. Cancer Cell 2013, 23, 489–501. [Google Scholar] [CrossRef]
- Kappert, F.; Sreeramulu, S.; Jonker, H.R.A.; Richter, C.; Rogov, V.V.; Proschak, E.; Hargittay, B.; Saxena, K.; Schwalbe, H. Structural Characterization of the Interaction of the Fibroblast Growth Factor Receptor with a Small Molecule Allosteric Inhibitor. Chem.—Eur. J. 2018, 24, 7861–7865. [Google Scholar] [CrossRef]
- Purslow, J.A.; Khatiwada, B.; Bayro, M.J.; Venditti, V. NMR Methods for Structural Characterization of Protein-Protein Complexes. Front. Mol. Biosci. 2020, 7, 9. [Google Scholar] [CrossRef]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and Allosteric Inhibition of the FGF2/HSPGs/FGFR1 Ternary Complex Formation by an Antiangiogenic, Thrombospondin-1-Mimic Small Molecule. PLoS ONE 2012, 7, e36990. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Foglieni, C.; Pagano, K.; Lessi, M.; Bugatti, A.; Moroni, E.; Pinessi, D.; Resovi, A.; Ribatti, D.; Bertini, S.; Ragona, L.; et al. Integrating computational and chemical biology tools in the discovery of antiangiogenic small molecule ligands of FGF2 derived from endogenous inhibitors. Sci. Rep. 2016, 6, 23432. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Carminati, L.; Tomaselli, S.; Molinari, H.; Taraboletti, G.; Ragona, L. Molecular Basis of the Antiangiogenic Action of Rosmarinic Acid, a Natural Compound Targeting Fibroblast Growth Factor-2/FGFR Interactions. ChemBioChem 2021, 22, 160–169. [Google Scholar] [CrossRef]
- Colombo, G.; Margosio, B.; Ragona, L.; Neves, M.; Bonifacio, S.; Annis, D.S.; Stravalaci, M.; Tomaselli, S.; Giavazzi, R.; Rusnati, M.; et al. Non-peptidic thrombospondin-1 mimics as fibroblast growth factor-2 inhibitors: An integrated strategy for the development of new antiangiogenic compounds. J. Biol. Chem. 2010, 285, 8733–8742. [Google Scholar] [CrossRef] [PubMed]
- Arablou, T.; Aryaeian, N.; Khodaverdi, S.; Kolahdouz-Mohammadi, R.; Moradi, Z.; Rashidi, N.; Delbandi, A.-A. The effects of resveratrol on the expression of VEGF, TGF-β, and MMP-9 in endometrial stromal cells of women with endometriosis. Sci. Rep. 2021, 11, 6054. [Google Scholar] [CrossRef] [PubMed]
- Bråkenhielm, E.; Cao, R.; Cao, Y. Suppression of angiogenesis, tumor growth, and wound healing by resveratrol, a natural compound in red wine and grapes. FASEB J. 2001, 15, 1798–1800. [Google Scholar] [CrossRef] [PubMed]
- Arablou, T.; Delbandi, A.A.; Khodaverdi, S.; Arefi, S.; Kolahdouz-Mohammadi, R.; Heidari, S.; Mohammadi, T.; Aryaeian, N. Resveratrol reduces the expression of insulin-like growth factor-1 and hepatocyte growth factor in stromal cells of women with endometriosis compared with nonendometriotic women. Phytotherapy Res. 2019, 33, 1044–1054. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef]
- Shimazaki, T.; Noro, N.; Hagikura, K.; Matsumoto, T.; Yoshida-Noro, C. Quantitative Analysis of Factors Regulating Angiogenesis for Stem Cell Therapy. Biology 2021, 10, 1212. [Google Scholar] [CrossRef]
- García-Caballero, M.; Marí-Beffa, M.; Cañedo, L.; Medina, M.Á.; Quesada, A.R. Toluquinol, a marine fungus metabolite, is a new angiosuppresor that interferes the Akt pathway. Biochem. Pharmacol. 2013, 85, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.S.; Cuevas, P.; Angulo, J.; López-Navajas, P.; Canales-Mayordomo, Á.; González-Corrochano, R.; Lozano, R.M.; Valverde, S.; Jiménez-Barbero, J.; Romero, A.; et al. Gentisic Acid, a Compound Associated with Plant Defense and a Metabolite of Aspirin, Heads a New Class of in Vivo Fibroblast Growth Factor Inhibitors. J. Biol. Chem. 2010, 285, 11714–11729. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, P.; Manquillo, A.; Guillen, P.; Giménez-Gallego, G. Fibroblast growth factor: A target for COVID-19 infection. Int. J. Med. Rev. Case Rep. 2020, 4, 122. [Google Scholar] [CrossRef]
- Elshorst, B.; Saxena, K.; Schieborr, U.; Schwalbe, H. 1H, 13C and 15N assignment of D2 domain of human fibroblast growth factor receptor 4. Biomol. NMR Assign. 2013, 7, 179–182. [Google Scholar] [CrossRef]
- Einstein, A. Investigations on the Theory of Brownian Movement; Dover Publications: Mineola, NY, USA, 1956. [Google Scholar]
- Jiménez-Martínez, T.S.; Romero–Manig, S.; Esturau-Escofet, N.; Briseño-Terán, M. DOSY Experiments to Monitor Block Copolymer Polymerization. J. Mex. Chem. Soc. 2011, 55, 4. [Google Scholar]
- Eliseo, T.; Ragona, L.; Catalano, M.; Assfalg, M.; Paci, M.; Zetta, L.; Molinari, H.; Cicero, D.O. Structural and dynamic determinants of ligand binding in the ternary complex of chicken liver bile acid binding protein with two bile salts revealed by NMR. Biochemistry 2007, 46, 12557–12567. [Google Scholar] [CrossRef]
- Stockman, B.J.; Dalvit, C. NMR screening techniques in drug discovery and drug design. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41, 187–231. [Google Scholar] [CrossRef]
- Cussler, E.L. Diffusion Mass Transfer in Fluid Systems, 2nd ed.; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Bocian, W.; Naumczuk, B.; Urbanowicz, M.; Sitkowski, J.; Bierczyńska-Krzysik, A.; Bednarek, E.; Wiktorska, K.; Milczarek, M.; Kozerski, L. The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons. Int. J. Mol. Sci. 2021, 22, 7471. [Google Scholar] [CrossRef]
- Moy, F.J.; Seddon, A.P.; Böhlen, P.; Powers, R. High-Resolution Solution Structure of Basic Fibroblast Growth Factor Determined by Multidimensional Heteronuclear Magnetic Resonance Spectroscopy. Biochemistry 1996, 35, 13552–13561. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Mulder, F.A.; Schipper, D.; Bott, R.; Boelens, R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999, 292, 111–123. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | D (×1010 m2/s) | Complexed Fraction |
|---|---|---|
| D2:FGF2 | 1.11 ± 0.02 1 | 0.39 2 |
| D2:FGF2:RA 3 | 1.19 ± 0.02 | 0.04 |
| D2:FGF2:RES 3 | 1.17 ± 0.02 | 0.12 |
| D2:FGF2:SM27 3 | 1.10 ± 0.02 | 0.42 |
| D2:FGF2:DOBE 3 | 1.12 ± 0.02 | 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, K.; Longhi, E.; Molinari, H.; Taraboletti, G.; Ragona, L. Inhibition of FGFR Signaling by Targeting FGF/FGFR Extracellular Interactions: Towards the Comprehension of the Molecular Mechanism through NMR Approaches. Int. J. Mol. Sci. 2022, 23, 10860. https://doi.org/10.3390/ijms231810860
Pagano K, Longhi E, Molinari H, Taraboletti G, Ragona L. Inhibition of FGFR Signaling by Targeting FGF/FGFR Extracellular Interactions: Towards the Comprehension of the Molecular Mechanism through NMR Approaches. International Journal of Molecular Sciences. 2022; 23(18):10860. https://doi.org/10.3390/ijms231810860
Chicago/Turabian StylePagano, Katiuscia, Elisa Longhi, Henriette Molinari, Giulia Taraboletti, and Laura Ragona. 2022. "Inhibition of FGFR Signaling by Targeting FGF/FGFR Extracellular Interactions: Towards the Comprehension of the Molecular Mechanism through NMR Approaches" International Journal of Molecular Sciences 23, no. 18: 10860. https://doi.org/10.3390/ijms231810860
APA StylePagano, K., Longhi, E., Molinari, H., Taraboletti, G., & Ragona, L. (2022). Inhibition of FGFR Signaling by Targeting FGF/FGFR Extracellular Interactions: Towards the Comprehension of the Molecular Mechanism through NMR Approaches. International Journal of Molecular Sciences, 23(18), 10860. https://doi.org/10.3390/ijms231810860