
MLK3 Regulates Inflammatory Response via Activation of AP-1 Pathway in HEK293 and RAW264.7 Cells



Abstract
:1. Introduction
2. Results
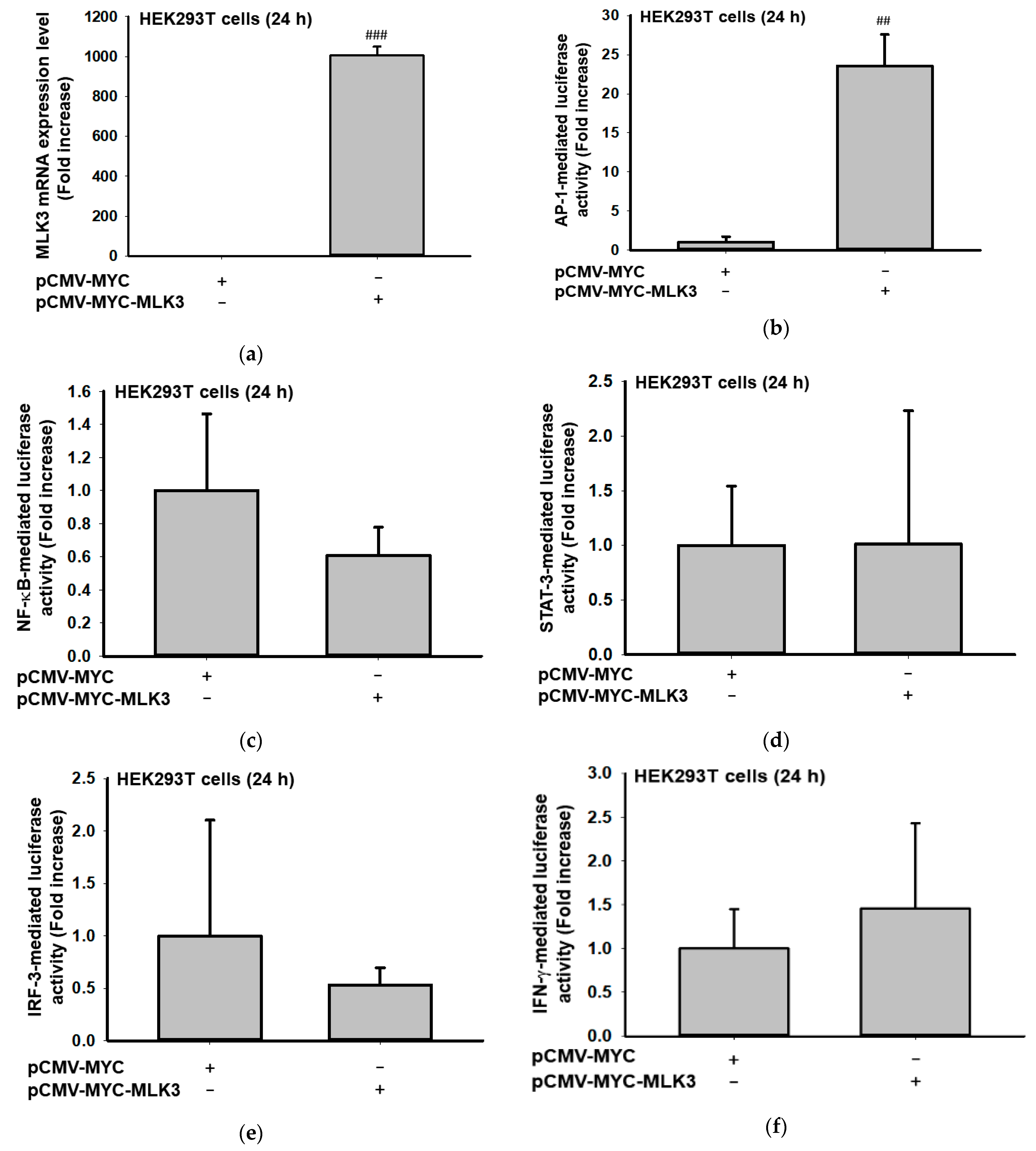
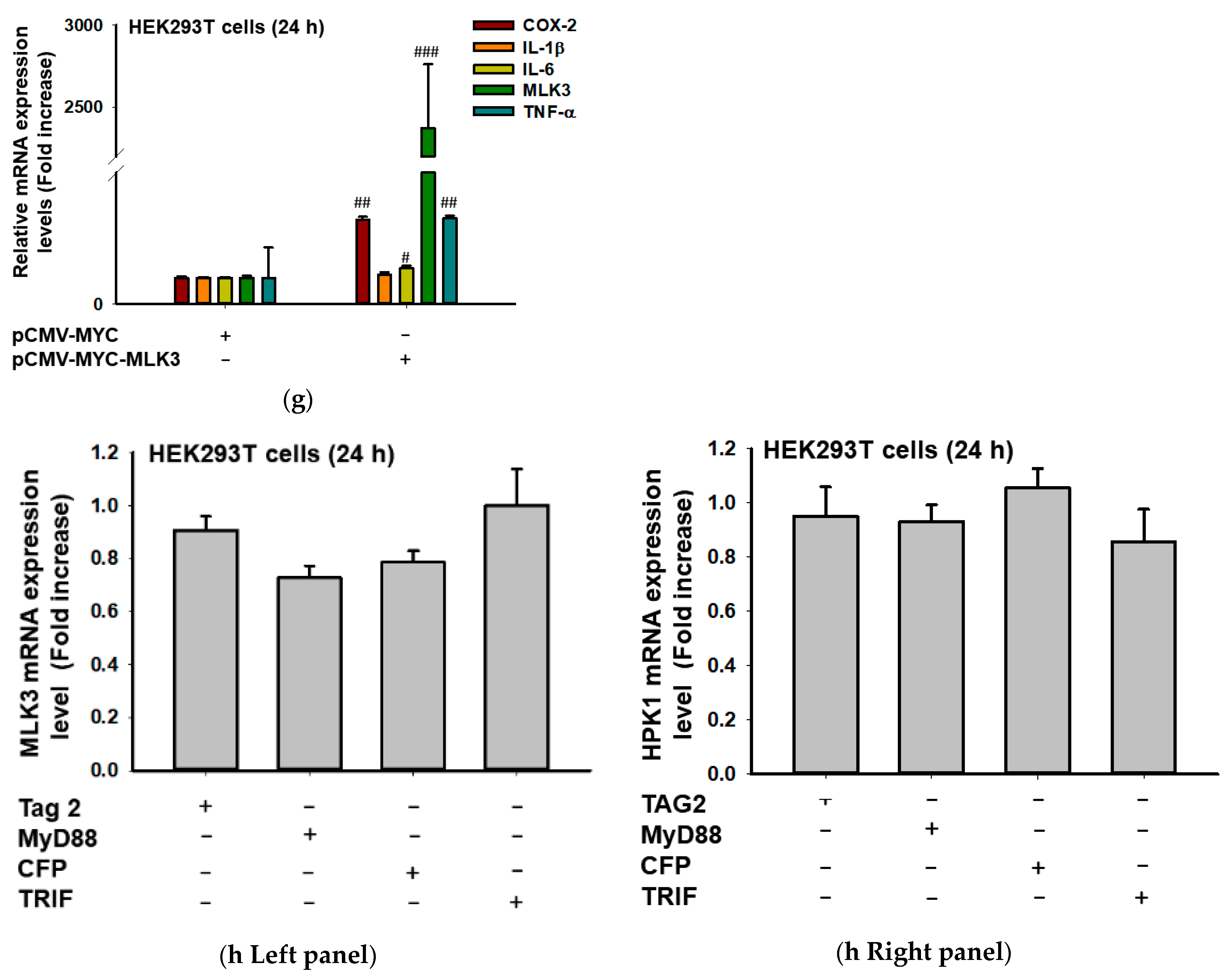
2.1. MLK3 Can Induce Expression of Inflammatory Genes via Activation of AP-1
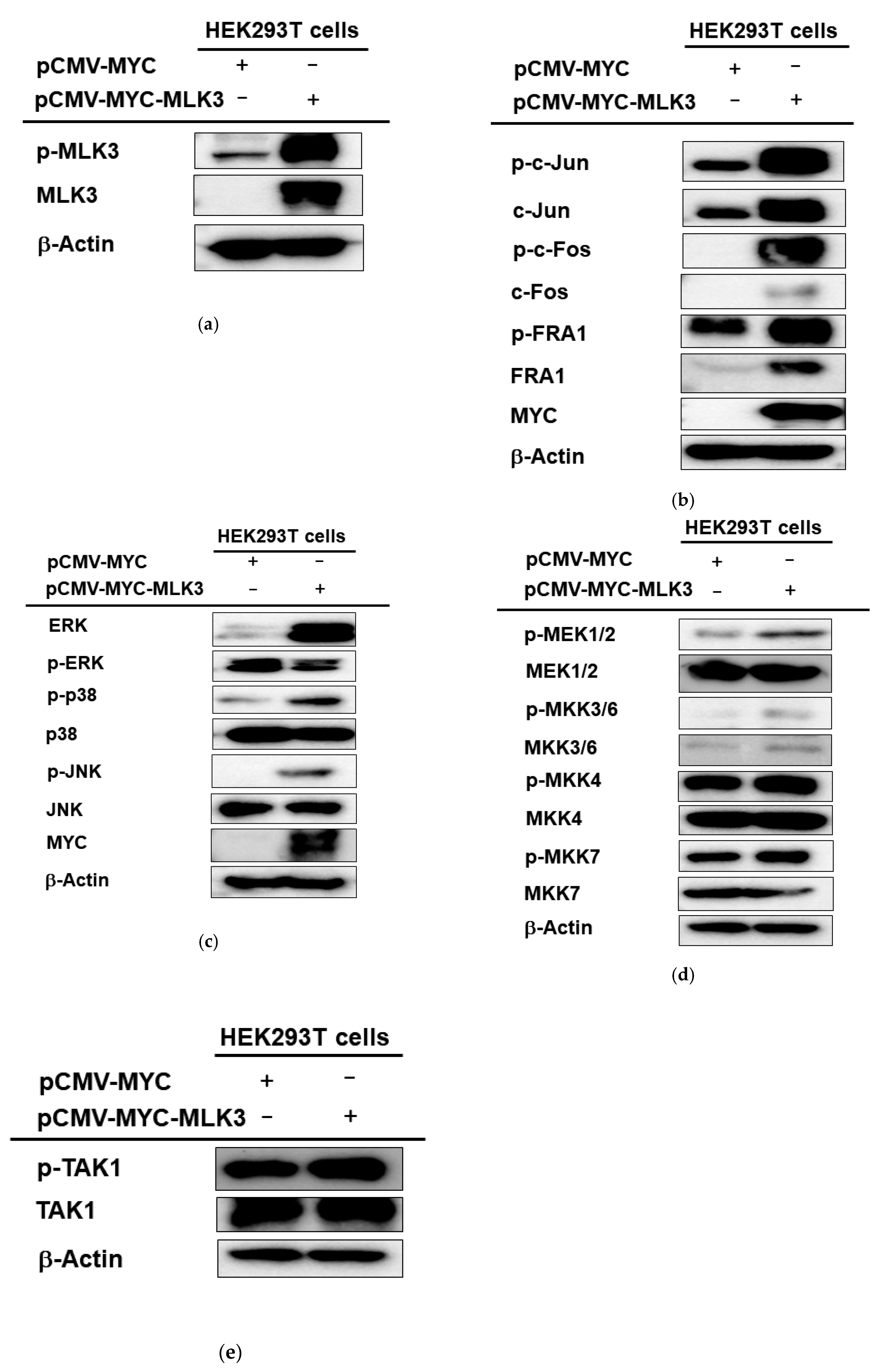
2.2. MLK3 Can Induce Phosphorylation of MAPKK, MAPK, and AP-1 Subunits
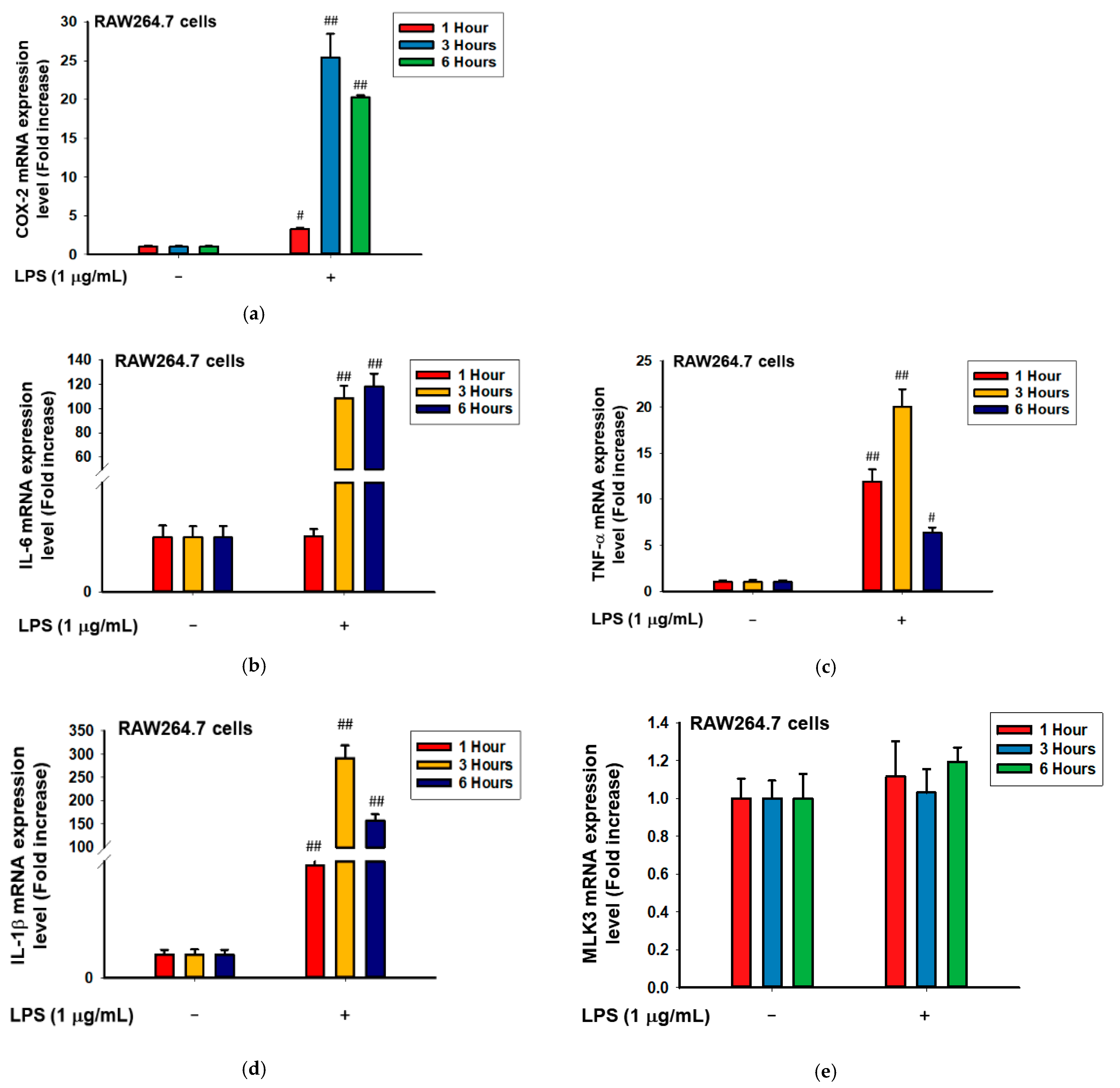
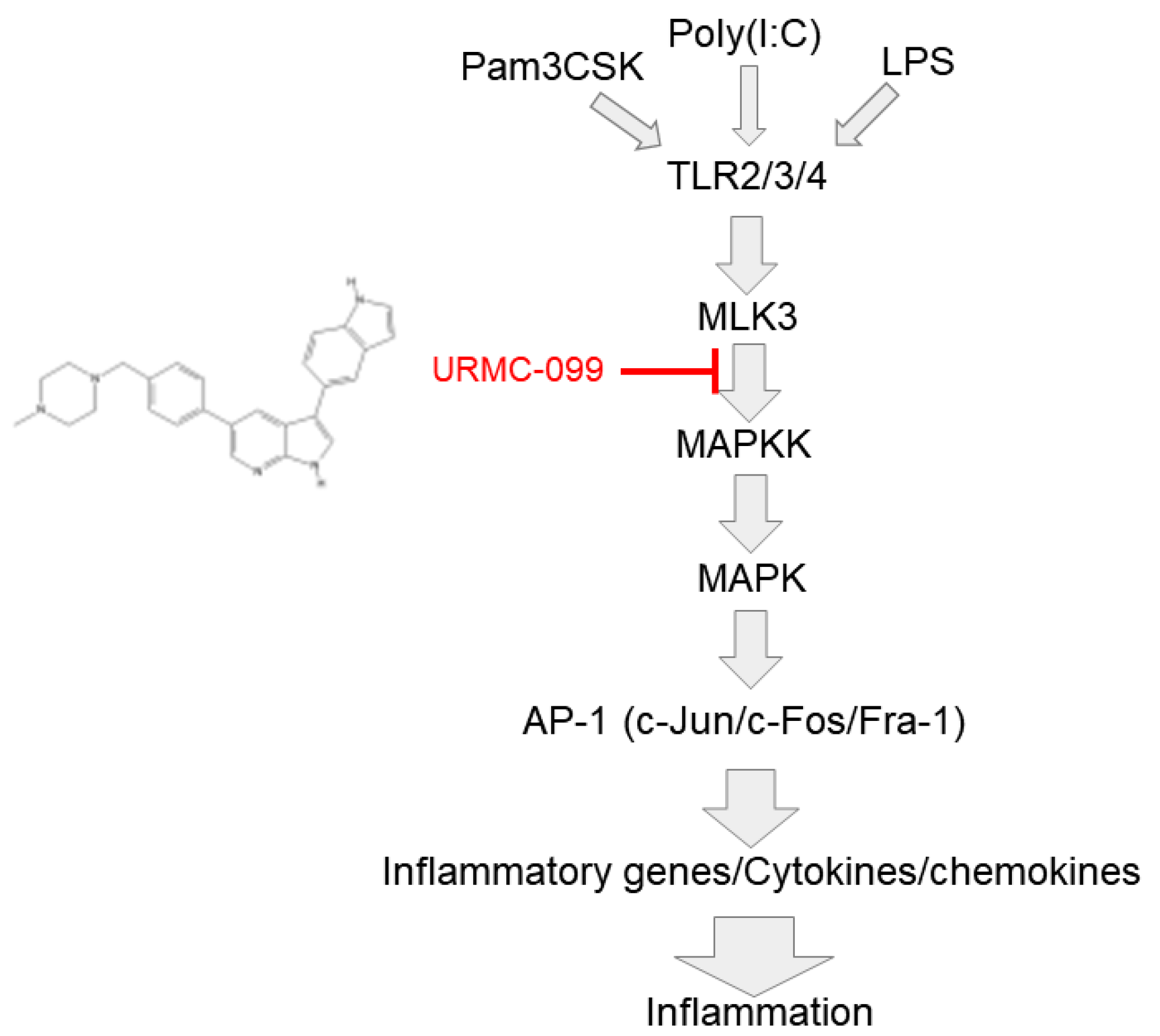
2.3. MLK3 Can Be Activated in RAW264.7 Cells Stimulated by LPS, Pam3CSK, and Poly(I:C)
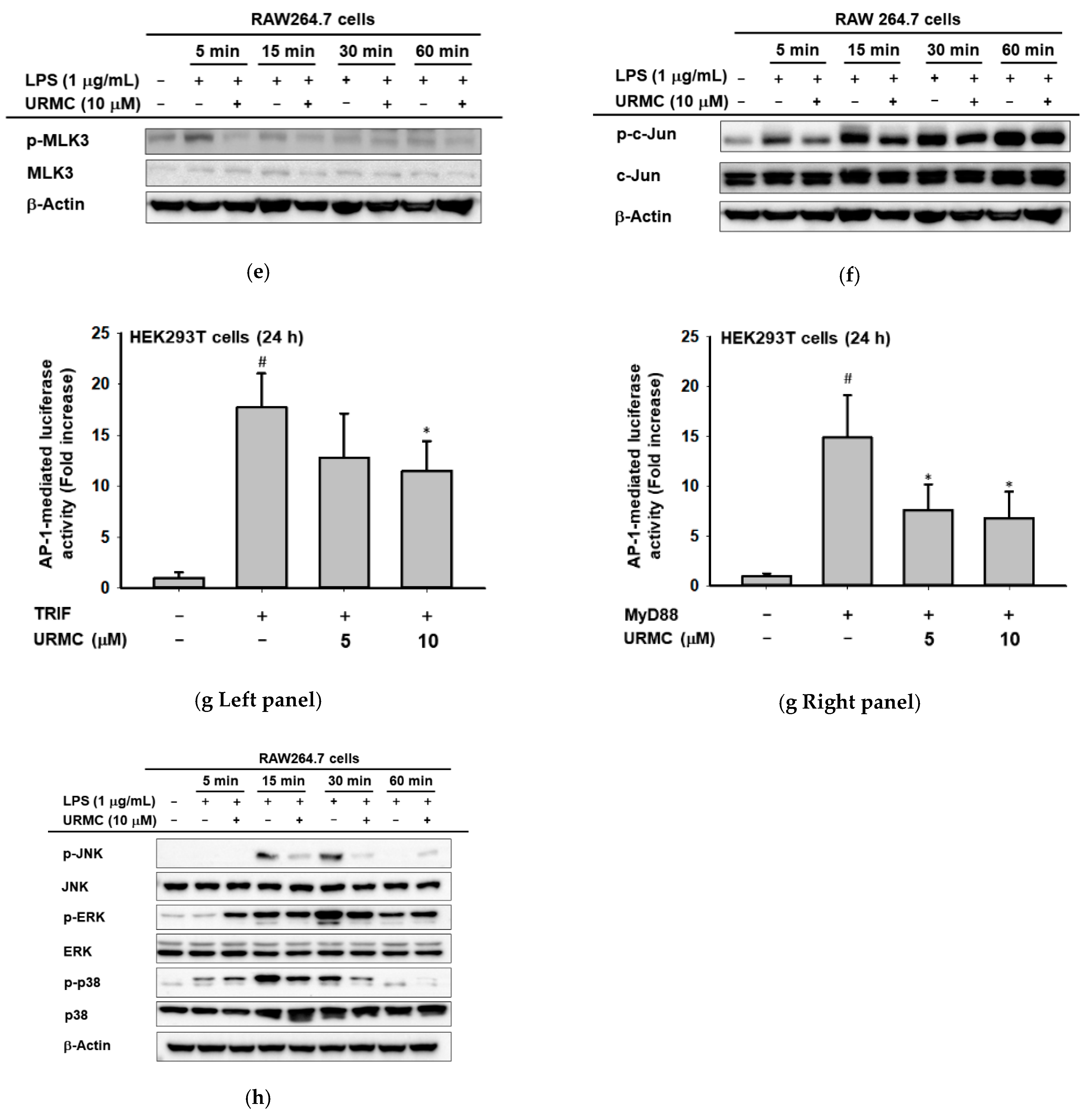
2.4. Inhibition of MLK3 Blocks AP-1-Mediated Inflammatory Response in LPS-Treated RAW264.7 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Cell Viability Tests
4.4. mRNA Analysis by Semi-Quantitative RT-PCR and Quantitative Real-Time PCR
4.5. Immunoblotting Analysis
4.6. Luciferase Reporter Gene Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| MLK3 | Mixed lineage kinase 3 |
| AP-1 | Activator protein-1 |
| LPS | Lipopolysaccharide |
| MAP3K | Mitogen-activated protein kinase kinase kinase |
| COX-2 | Cyclooxygenase-2 |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| DAMP | Damage-associated molecular patterns |
| TLR | Toll-like receptors |
| MyD88 | Myeloid differentiation factor 88 |
| TRIF | TIR-domain-containing adaptor-inducing interferon-β |
| MAPKs | Mitogen-activated protein kinases |
| TAK-1 | Transforming growth factor-β-activated kinase 1 |
| TNF-α | Tumor necrosis factor-α |
| NF-κB | Nuclear factor-κB |
| IRF3 | Interferon regulatory factor 3 |
| STAT-3 | Signal transducer and activator of transcription 3 |
| PEI | Polyethylenimine |
| PRR | pattern recognition receptor |
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, X.H.; Li, J.R.; Zheng, T.H.; Yao, F.B.; Gao, B.; Xue, T.C. Neutrophils: Driving inflammation during the development of hepatocellular carcinoma. Cancer Lett. 2021, 522, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yuan, D.; Gai, L.; Wu, X.; Shi, Y.; He, Y.; Liu, C.; Zhang, C.; Zhou, G.; Yuan, C. Saponins from Panax japonicus ameliorate age-related renal fibrosis by inhibition of inflammation mediated by NF-kappaB and TGF-beta1/Smad signaling and suppression of oxidative stress via activation of Nrf2-ARE signaling. J. Ginseng Res. 2021, 45, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal os that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, H.G.; Kim, E.; Han, S.Y.; Aziz, N.; Yi, Y.S.; Kim, S.; Lee, Y.; Yoo, B.C.; Han, J.W.; et al. Isoprenylcysteine carboxyl methyltransferase and its substrate Ras are critical players regulating TLR-mediated inflammatory responses. Cells 2020, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.J.S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S.J.C. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Ranjbar, A.; Seyed Saleh, S.H.; Bagherian, M.; Sharifzadeh, S.O.; Hushmandi, K.; et al. Regulation of Nuclear Factor-KappaB (NF-kappaB) signaling pathway by non-coding RNAs in cancer: Inhibiting or promoting carcinogenesis? Cancer Lett. 2021, 509, 63–80. [Google Scholar] [CrossRef]
- Kim, S.A.; Lee, C.Y.; Mitra, A.; Kim, H.; Woo, B.Y.; Hong, Y.D.; Noh, J.K.; Yi, D.K.; Kim, H.G.; Cho, J.Y. Anti-inflammatory effects of Huberia peruviana Cogn. methanol extract by inhibiting Src activity in the NF-kappaB pathway. Plants 2021, 10, 2335. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Pudelko, K.; Biermeier, A.; Walterskirchen, N.; Gaigneaux, A.; Weindorfer, C.; Harrer, N.; Klett, H.; Hengstschlager, M.; Schuler, J.; et al. Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett. 2021, 520, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Sun, Z.; Zhan, H. Macrophages in pancreatic cancer: An immunometabolic perspective. Cancer Lett. 2021, 498, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; He, J.; Dong, J.; Wu, S.; Liu, S.; Zhu, H.; Xu, T. UM-6 induces autophagy and apoptosis via the Hippo-YAP signaling pathway in cervical cancer. Cancer Lett. 2021, 519, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Liu, S.; Zhang, J.; Pei, M.; Xiao, Y.; Li, J.; Hong, L.; Lin, J.; Wang, J.; Wu, X.; et al. Vorinostat triggers miR-769-5p/3p-mediated suppression of proliferation and induces apoptosis via the STAT3-IGF1R-HDAC3 complex in human gastric cancer. Cancer Lett. 2021, 521, 196–209. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wagner, E.F. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 2011, 70, i109–i112. [Google Scholar] [CrossRef]
- Liu, X.; Yin, S.; Chen, Y.; Wu, Y.; Zheng, W.; Dong, H.; Bai, Y.; Qin, Y.; Li, J.; Feng, S.J.M. LPS-induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF-κB, STAT3 or AP-1 activation. Mol. Med. Rep. 2018, 17, 5484–5491. [Google Scholar] [CrossRef]
- Song, D.; He, H.; Sinha, I.; Hases, L.; Yan, F.; Archer, A.; Haldosen, L.A.; Zhao, C.; Williams, C. Blocking Fra-1 sensitizes triple-negative breast cancer to PARP inhibitor. Cancer Lett. 2021, 506, 23–34. [Google Scholar] [CrossRef]
- Yang, X.; Shao, C.; Duan, L.; Hou, X.; Huang, Y.; Gao, L.; Zong, C.; Liu, W.; Jiang, J.; Ye, F.; et al. Oncostatin M promotes hepatic progenitor cell activation and hepatocarcinogenesis via macrophage-derived tumor necrosis factor-alpha. Cancer Lett. 2021, 517, 46–54. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Z.; She, Y.; Deng, J.; Zhong, Y.; Zhao, M.; Li, S.; Xie, D.; Sun, X.; Hu, X.; et al. A novel protein encoded by circASK1 ameliorates gefitinib resistance in lung adenocarcinoma by competitively activating ASK1-dependent apoptosis. Cancer Lett. 2021, 520, 321–331. [Google Scholar] [CrossRef]
- Ma, Q.; Xu, Q.; Zhao, J.; Zhang, W.; Wang, Q.; Fang, J.; Lu, Z.; Liu, J.; Ma, L. Coupling HDAC4 with transcriptional factor MEF2D abrogates SPRY4-mediated suppression of ERK activation and elicits hepatocellular carcinoma drug resistance. Cancer Lett. 2021, 520, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Kajanne, R.; Miettinen, P.; Mehlem, A.; Leivonen, S.K.; Birrer, M.; Foschi, M.; Kähäri, V.M.; Leppä, S. EGF-R regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell. Physiol. 2007, 212, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-W.; Park, K.; Kweon, G.R.; Jang, B.-C.; Baek, W.-K.; Suh, M.-H.; Kim, C.-W.; Lee, K.-S.; Suh, S.-I. Curcumin inhibits the expression of COX-2 in UVB-irradiated human keratinocytes (HaCaT) by inhibiting activation of AP-1: p38 MAP kinase and JNK as potential upstream targets. Exp. Mol. Med. 2005, 37, 186–192. [Google Scholar] [CrossRef]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.; Pischke, S.E.; Zhou, Z.; Davis, R.J.; Flavell, R.A.; Loop, T.; Otterbein, S.L.; Otterbein, L.E.; Choi, A.M. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 2003, 278, 36993–36998. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, S.K.; Rana, B.; Rana, A. The regulatory function of mixed lineage kinase 3 in tumor and host immunity. Pharmacol. Ther. 2021, 219, 107704. [Google Scholar] [CrossRef]
- Viswakarma, N.; Sondarva, G.; Principe, D.R.; Nair, R.S.; Kumar, S.; Singh, S.K.; Das, S.; Sinha, S.C.; Grippo, P.J.; Grimaldo, S.; et al. Mixed Lineage Kinase 3 phosphorylates prolyl-isomerase PIN1 and potentiates GLI1 signaling in pancreatic cancer development. Cancer Lett. 2021, 515, 1–13. [Google Scholar] [CrossRef]
- Handley, M.E.; Rasaiyaah, J.; Chain, B.M.; Katz, D.R. Mixed lineage kinases (MLKs): A role in dendritic cells, inflammation and immunity? Int. J. Exp. Pathol. 2007, 88, 111–126. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, Y.; Wu, S.; Hou, G.; Wu, N.; Qian, L.; Yang, D. MLK3 is a newly identified microRNA-520b target that regulates liver cancer cell migration. PLoS ONE 2020, 15, e0230716. [Google Scholar] [CrossRef]
- Kasturirangan, S.; Mehdi, B.; Chadee, D.N. LATS1 regulates mixed-lineage kinase 3 (MLK3) subcellular localization and MLK3-mediated invasion in ovarian epithelial cells. Mol. Cell Biol. 2021, 41, e0007821. [Google Scholar] [CrossRef]
- Das, S.; Nair, R.S.; Mishra, R.; Sondarva, G.; Viswakarma, N.; Abdelkarim, H.; Gaponenko, V.; Rana, B.; Rana, A. Mixed lineage kinase 3 promotes breast tumorigenesis via phosphorylation and activation of p21-activated kinase 1. Oncogene 2019, 38, 3569–3584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a double-edge sword for cancer and other age-related diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L.J.O. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Duan, M.; Zheng, F.; Yu, L.; Wang, Y.; Wang, G.; Lin, J.; Han, S.; Gan, D.; Meng, Z.; et al. Fas signaling in adipocytes promotes low-grade inflammation and lung metastasis of colorectal cancer through interaction with Bmx. Cancer Lett. 2021, 522, 93–104. [Google Scholar] [CrossRef]
- Xue, Q.; He, N.; Wang, Z.; Fu, X.; Aung, L.H.H.; Liu, Y.; Li, M.; Cho, J.Y.; Yang, Y.; Yu, T. Functional roles and mechanisms of ginsenosides from Panax ginseng in atherosclerosis. J. Ginseng Res. 2021, 45, 22–31. [Google Scholar] [CrossRef]
- Liu, M.; Qin, Y.; Hu, Q.; Liu, W.; Ji, S.; Xu, W.; Fan, G.; Ye, Z.; Zhang, Z.; Xu, X.; et al. SETD8 potentiates constitutive ERK1/2 activation via epigenetically silencing DUSP10 expression in pancreatic cancer. Cancer Lett. 2021, 499, 265–278. [Google Scholar] [CrossRef]
- Cho, J.Y.; Grigura, V.; Murphy, T.L.; Murphy, K. Identification of cooperative monomeric Brachyury sites conferring T-bet responsiveness to the proximal IFN-gamma promoter. Int. Immunol. 2003, 15, 1149–1160. [Google Scholar] [CrossRef]
- Arnold, R.; Frey, C.R.; Muller, W.; Brenner, D.; Krammer, P.H.; Kiefer, F. Sustained JNK signaling by proteolytically processed HPK1 mediates IL-3 independent survival during monocytic differentiation. Cell Death Differ. 2007, 14, 568–575. [Google Scholar] [CrossRef]
- Schroyer, A.L.; Stimes, N.W.; Abi Saab, W.F.; Chadee, D.N. MLK3 phosphorylation by ERK1/2 is required for oxidative stress-induced invasion of colorectal cancer cells. Oncogene 2018, 37, 1031–1040. [Google Scholar] [CrossRef]
- Goodfellow, V.S.; Loweth, C.J.; Ravula, S.B.; Wiemann, T.; Nguyen, T.; Xu, Y.; Todd, D.E.; Sheppard, D.; Pollack, S.; Polesskaya, O.; et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3. J. Med. Chem. 2013, 56, 8032–8048. [Google Scholar] [CrossRef]
- Miller-Rhodes, P.; Kong, C.; Baht, G.S.; Saminathan, P.; Rodriguiz, R.M.; Wetsel, W.C.; Gelbard, H.A.; Terrando, N. The broad spectrum mixed-lineage kinase 3 inhibitor URMC-099 prevents acute microgliosis and cognitive decline in a mouse model of perioperative neurocognitive disorders. J. Neuroinflamm. 2019, 16, 193. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, M.J.; Hammond, J.W.; Li, H.; Gantz Marker, M.A.; Marker, D.F.; Freeman, R.S.; Gelbard, H.A. The mixed-lineage kinase inhibitor URMC-099 protects hippocampal synapses in experimental autoimmune encephalomyelitis. eNeuro 2018, 5, ENEURO.0245-18.2018. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Zhang, G.; Mosley, R.L.; Gelbard, H.A.; Gendelman, H.E. URMC-099 facilitates amyloid-beta clearance in a murine model of Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 137. [Google Scholar] [CrossRef] [PubMed]
- Rhoo, K.H.; Granger, M.; Sur, J.; Feng, C.; Gelbard, H.A.; Dewhurst, S.; Polesskaya, O. Pharmacologic inhibition of MLK3 kinase activity blocks the in vitro migratory capacity of breast cancer cells but has no effect on breast cancer brain metastasis in a mouse xenograft model. PLoS ONE 2014, 9, e108487. [Google Scholar] [CrossRef]
- Polesskaya, O.; Wong, C.; Lebron, L.; Chamberlain, J.M.; Gelbard, H.A.; Goodfellow, V.; Kim, M.; Daiss, J.L.; Dewhurst, S. MLK3 regulates fMLP-stimulated neutrophil motility. Mol. Immunol. 2014, 58, 214–222. [Google Scholar] [CrossRef]
- Kim, H.G.; Lee, C.; Yoon, J.H.; Kim, J.H.; Cho, J.Y. BN82002 alleviated tissue damage of septic mice by reducing inflammatory response through inhibiting AKT2/NF-kappaB signaling pathway. Biomed. Pharmacother. 2022, 148, 112740. [Google Scholar] [CrossRef]
- Park, J.G.; Yi, Y.S.; Hong, Y.H.; Yoo, S.; Han, S.Y.; Kim, E.; Jeong, S.G.; Aravinthan, A.; Baik, K.S.; Choi, S.Y.; et al. Tabetri (Tabebuia avellanedae ethanol extract) ameliorates osteoarthritis symptoms induced by monoiodoacetate through its anti-inflammatory and chondroprotective activities. Mediat. Inflamm. 2017, 2017, 3619879. [Google Scholar] [CrossRef]
- Kim, S.A.; Oh, J.; Choi, S.R.; Lee, C.H.; Lee, B.H.; Lee, M.N.; Hossain, M.A.; Kim, J.H.; Lee, S.; Cho, J.Y. Anti-gastritis and anti-lung injury effects of pine tree ethanol extract targeting both NF-kappaB and AP-1 pathways. Molecules 2021, 26, 6275. [Google Scholar] [CrossRef]
- Lee, J.O.; Hwang, S.H.; Shen, T.; Kim, J.H.; You, L.; Hu, W.; Cho, J.Y. Enhancement of skin barrier and hydration-related molecules by protopanaxatriol in human keratinocytes. J. Ginseng Res. 2021, 45, 354–360. [Google Scholar] [CrossRef]
- Jo, H.; Jang, D.; Park, S.K.; Lee, M.G.; Cha, B.; Park, C.; Shin, Y.S.; Park, H.; Baek, J.M.; Heo, H.; et al. Ginsenoside 20(S)-protopanaxadiol induces cell death in human endometrial cancer cells via apoptosis. J. Ginseng Res. 2021, 45, 126–133. [Google Scholar] [CrossRef]
- Lee, J.O.; Kim, J.H.; Kim, S.; Kim, M.Y.; Hong, Y.H.; Kim, H.G.; Cho, J.Y. Gastroprotective effects of the nonsaponin fraction of Korean Red Ginseng through cyclooxygenase-1 upregulation. J. Ginseng Res. 2020, 44, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Oh, J.; Jo, M.; Kim, J.K.; Kim, D.S.; Kim, H.G.; Yoon, K.; Yang, Y.; Geum, J.H.; Kim, J.E.; et al. Water extract of Lotus leaf alleviates dexamethasone-induced muscle atrophy via regulating protein metabolism-related pathways in mice. Molecules 2020, 25, 4592. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Aravinthan, A.; Hossain, M.A.; Kopalli, S.R.; Kim, B.; Kim, N.S.; Kang, C.W.; Kim, J.H. Effect of Korean Red Ginseng through comparative analysis of cardiac gene expression in db/db mice. J. Ginseng Res. 2021, 45, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Hwang, K.; Lee, J.; Han, S.Y.; Kim, E.M.; Park, J.; Cho, J.Y. Skin protective effect of epigallocatechin gallate. Int. J. Mol. Sci. 2018, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, C.J. Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignan, inhibits type I-IV allergic inflammation and pro-inflammatory enzymes. Arch. Pharm. Res. 2010, 33, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Devitt, G.; Thomas, M.; Klibanov, A.M.; Pfeiffer, T.; Bosch, V. Optimized protocol for the large scale production of HIV pseudovirions by transient transfection of HEK293T cells with linear fully deacylated polyethylenimine. J. Virol. Methods 2007, 146, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Tao, L.Y.; Yang, M.W.; Xu, D.P.; Jiang, S.H.; Fu, X.L.; Liu, D.J.; Huo, Y.M.; Liu, W.; Yang, J.Y.; et al. CD74 promotes perineural invasion of cancer cells and mediates neuroplasticity via the AKT/EGR-1/GDNF axis in pancreatic ductal adenocarcinoma. Cancer Lett. 2021, 508, 47–58. [Google Scholar] [CrossRef]
- Zhang, T.; Zhong, S.; Hou, L.; Wang, Y.; Xing, X.; Guan, T.; Zhang, J.; Li, T. Computational and experimental characterization of estrogenic activities of 20(S, R)-protopanaxadiol and 20(S, R)-protopanaxatriol. J. Ginseng Res. 2020, 44, 690–696. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Direction | Sequence (5′ to 3′) |
|---|---|---|
| Primer Sequences used in RT-real-time PCR | ||
| COX-2 | Forward | TTGGAGGCGAAGTGGGTTTT |
| Reverse | TGGCTGTTTTGGTAGGCTGT | |
| TNF-α | Forward | TGCCTATGTCTCAGCCTCTT |
| Reverse | GAGGCCATTTGGGAACTTCT | |
| IL-1β | Forward | GTGAAATGCCACCTTTTGACAGTG |
| Reverse | CCTGCCTGAAGCTCTTGTTG | |
| IL-6 | Forward | AGCCAGAGTCCTTCAGAGAGAT |
| Reverse | AGGAGAGCATTGGAAATTGGGG | |
| CCL-12 | Forward | GCCTCCTGCTCATAGCTACC |
| Reverse | CTTCCGGACGTGAATCTTCT | |
| MLK3 | Forward | GTCGACAATGGAGCCCTTGAAGAGCCTC |
| Reverse | CGGCCGTCAAGGCCCCGCTTCCG | |
| HPK1 | Forward | CTGCTGGAACGGAAAGAGAC |
| Reverse | CGGACAAGCAGGAATTTGTT | |
| GAPDH | Forward | TGTGAACGGATTTGGCCGTA |
| Reverse | ACTGTGCCGTTGAATTTGCC | |
| Primer Sequences used in RT-PCR | ||
| COX 2 | Forward | TCACGTGGAGTCCGCTTTAC |
| Reverse | CTTCGCAGGAAGGGGATGTT | |
| CCL-12 | Forward | GCCTCCTGCTCATAGCTACC |
| Reverse | CTTCCGGACGTGAATCTTCT | |
| GAPDH | Forward | CACTCACGGCAAATTCAACGGCA |
| Reverse | GACTCCACGACATACTCAGCAC | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, A.T.; Cho, J.Y.; Kim, D. MLK3 Regulates Inflammatory Response via Activation of AP-1 Pathway in HEK293 and RAW264.7 Cells. Int. J. Mol. Sci. 2022, 23, 10874. https://doi.org/10.3390/ijms231810874
Ha AT, Cho JY, Kim D. MLK3 Regulates Inflammatory Response via Activation of AP-1 Pathway in HEK293 and RAW264.7 Cells. International Journal of Molecular Sciences. 2022; 23(18):10874. https://doi.org/10.3390/ijms231810874
Chicago/Turabian StyleHa, Anh Thu, Jae Youl Cho, and Daewon Kim. 2022. "MLK3 Regulates Inflammatory Response via Activation of AP-1 Pathway in HEK293 and RAW264.7 Cells" International Journal of Molecular Sciences 23, no. 18: 10874. https://doi.org/10.3390/ijms231810874