

Cancer Resistance to Immunotherapy: Molecular Mechanisms and Tackling Strategies


Abstract
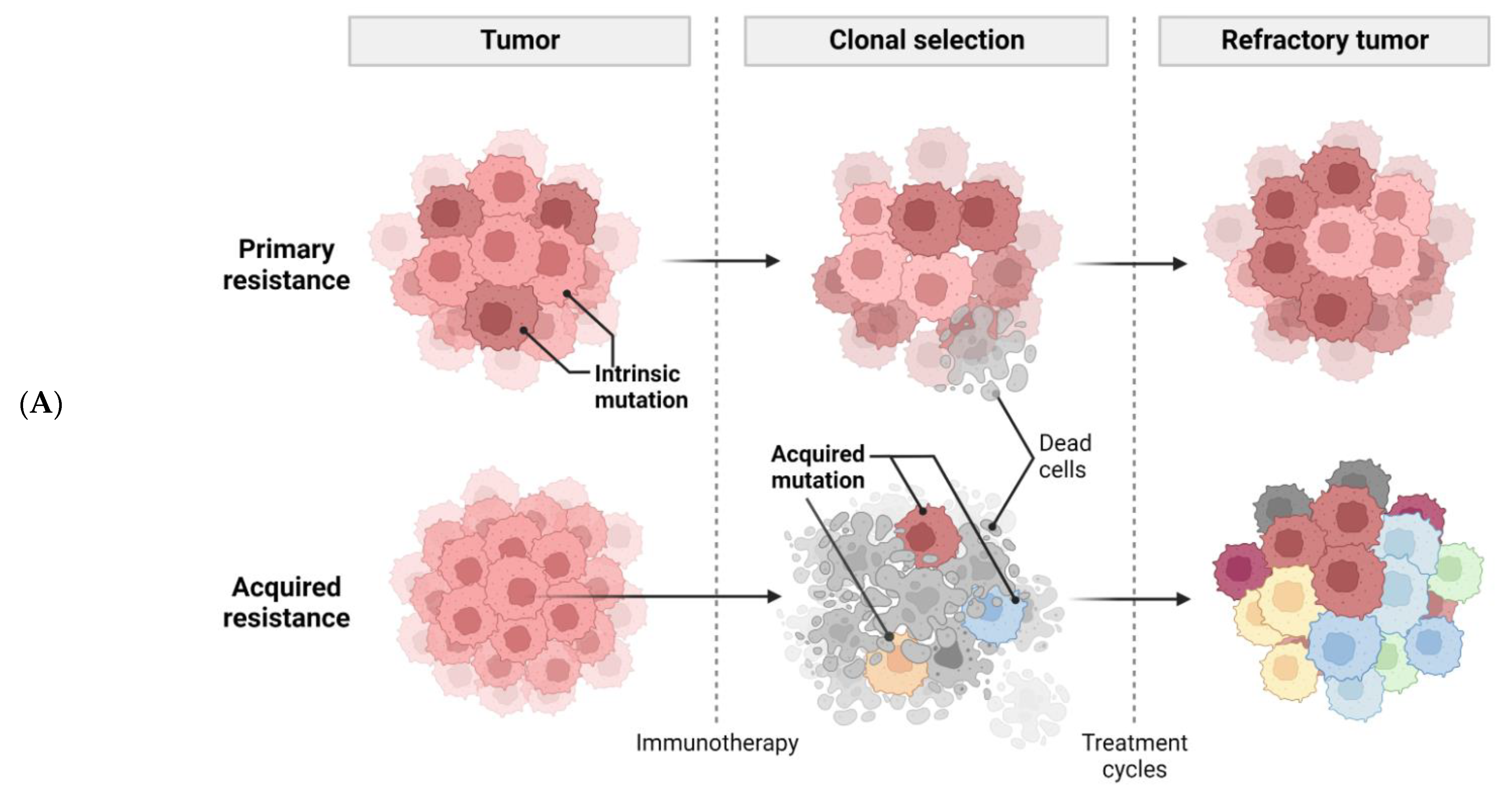
1. Introduction
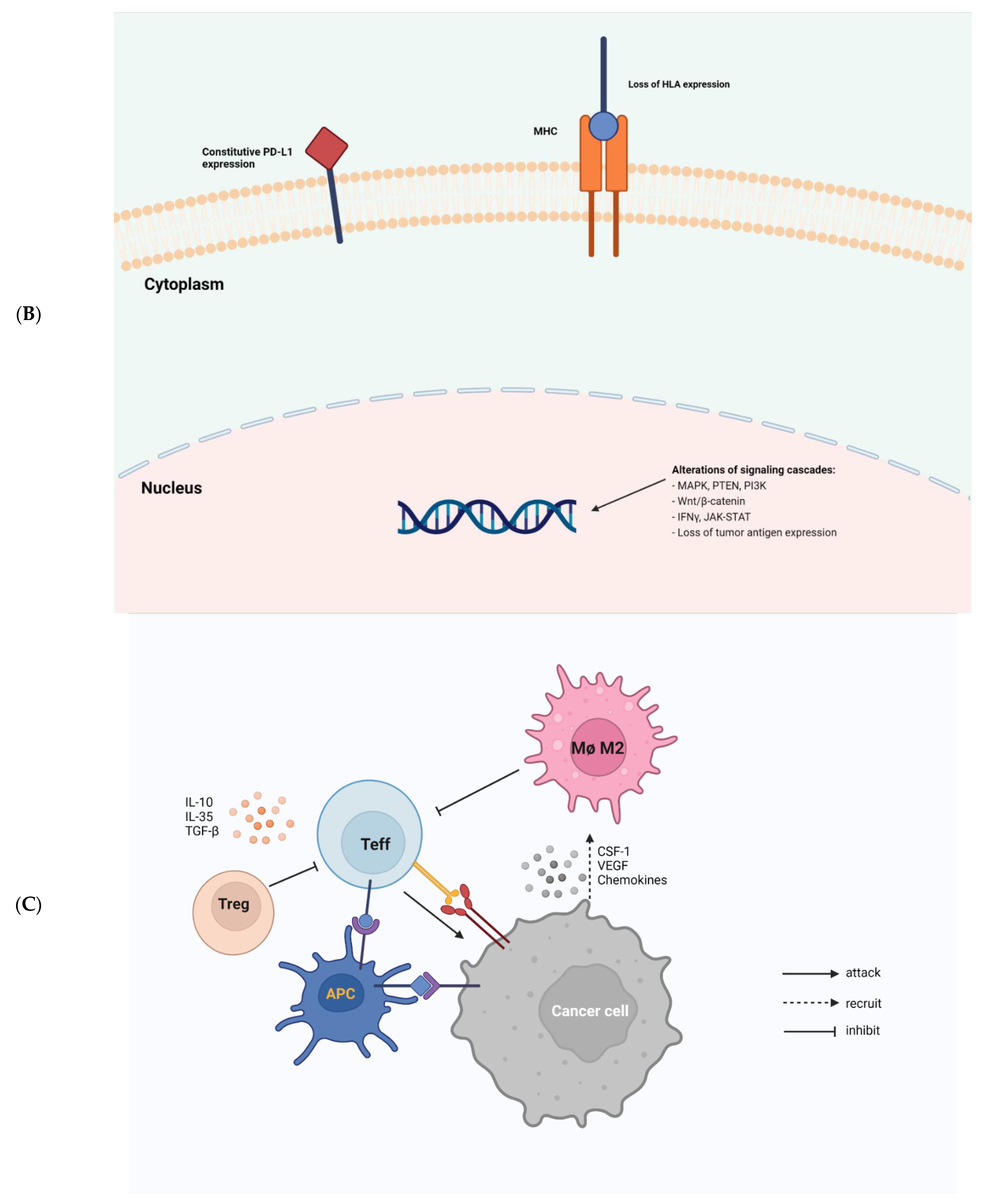
2. Molecular Mechanisms
2.1. Tumor Intrinsic Factors of Resistance to Immunotherapy
2.2. Tumor Extrinsic Factors of Resistance to Immunotherapy
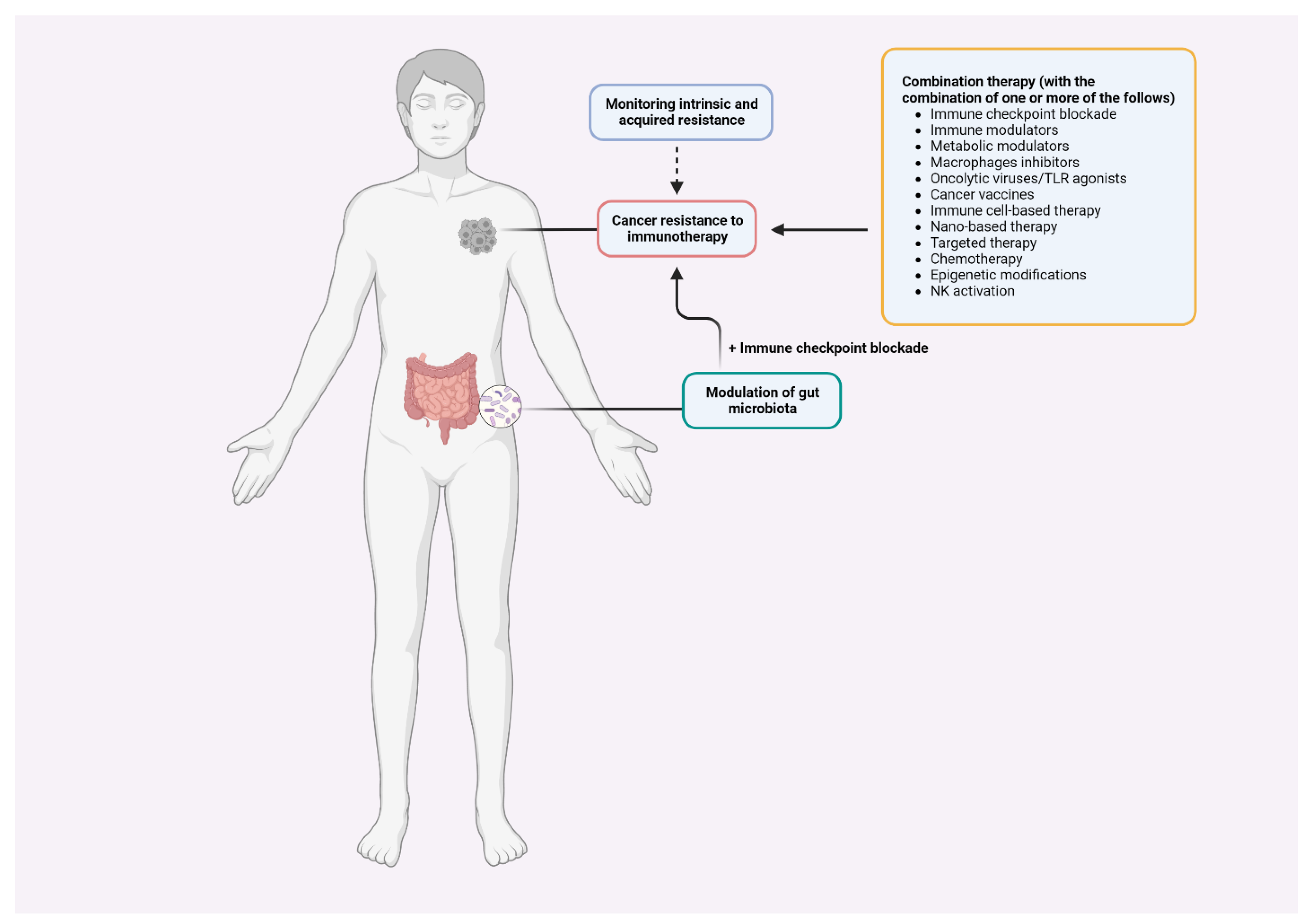
3. Tackling Strategies
4. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nature Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005, 17, 133–144. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef]
- Larkin, J.; Lao, C.D.; Urba, W.J.; McDermott, D.F.; Horak, C.; Jiang, J.; Wolchok, J.D. Efficacy and Safety of Nivolumab in Patients with BRAF V600 Mutant and BRAF Wild-Type Advanced Melanoma: A Pooled Analysis of 4 Clinical Trials. JAMA Oncol. 2015, 1, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Lizee, G.; Schueneman, A.J. Endogenous T-Cell Therapy: Clinical Experience. Cancer J. 2015, 21, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- June, C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Investig. 2007, 117, 1466–1476. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef]
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell Therapy for Cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [CrossRef]
- Sadelain, M. Chimeric antigen receptors: Driving immunology towards synthetic biology. Curr. Opin. Immunol. 2016, 41, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Kalbasi, A.; Siurala, M.; Su, L.L.; Tariveranmoshabad, M.; Picton, L.K.; Ravikumar, P.; Li, P.; Lin, J.-X.; Escuin-Ordinas, H.; Da, T.; et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature 2022, 607, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Maeurer, M.J.; Gollin, S.M.; Storkus, W.J.; Swaney, W.; Karbach, J.; Martin, D.; Castelli, C.; Salter, R.; Knuth, A.; Lotze, M.T. Tumor escape from immune recognition: Loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 641–652. [Google Scholar]
- Hazini, A.; Fisher, K.; Seymour, L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J. Immunother. Cancer 2021, 9, e002899. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef] [PubMed]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Garrido, F. MHC/HLA Class I Loss in Cancer Cells. Adv. Exp. Med. Biol. 2019, 1151, 15–78. [Google Scholar] [CrossRef]
- Tanaka, K.; Tsuchikawa, T.; Miyamoto, M.; Maki, T.; Ichinokawa, M.; Kubota, K.C.; Shichinohe, T.; Hirano, S.; Ferrone, S.; Dosaka-Akita, H.; et al. Down-regulation of Human Leukocyte Antigen class I heavy chain in tumors is associated with a poor prognosis in advanced esophageal cancer patients. Int. J. Oncol. 2012, 40, 965–974. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Roulois, D.; Yau, H.L.; De Carvalho, D.D. Pharmacological DNA demethylation: Implications for cancer immunotherapy. Oncoimmunology 2016, 5, e1090077. [Google Scholar] [CrossRef]
- Kirkin, A.F.; Dzhandzhugazyan, K.N.; Guldberg, P.; Fang, J.J.; Andersen, R.S.; Dahl, C.; Mortensen, J.; Lundby, T.; Wagner, A.; Law, I.; et al. Adoptive cancer immunotherapy using DNA-demethylated T helper cells as antigen-presenting cells. Nat. Commun. 2018, 9, 785. [Google Scholar] [CrossRef]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef]
- Rosborough, B.R.; Castellaneta, A.; Natarajan, S.; Thomson, A.W.; Turnquist, H.R. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J. Leukoc. Biol. 2012, 91, 701–709. [Google Scholar] [CrossRef]
- Wang, H.F.; Ning, F.; Liu, Z.C.; Wu, L.; Li, Z.Q.; Qi, Y.F.; Zhang, G.; Wang, H.S.; Cai, S.H.; Du, J. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. CII 2017, 66, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Sundstedt, A.; O’Neill, E.J.; Nicolson, K.S.; Wraith, D.C. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J. Immunol. 2003, 170, 1240–1248. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Oida, T.; Zhang, X.; Goto, M.; Hachimura, S.; Totsuka, M.; Kaminogawa, S.; Weiner, H.L. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J. Immunol. 2003, 170, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e644. [Google Scholar] [CrossRef]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Weber, A.; Sarnaik, A.A.; Pilon-Thomas, S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J. Immunol. 2012, 189, 5147–5154. [Google Scholar] [CrossRef]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. CII 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Hu, W.; Li, X.; Zhang, C.; Yang, Y.; Jiang, J.; Wu, C. Tumor-associated macrophages in cancers. Clin. Transl. Oncol. 2016, 18, 251–258. [Google Scholar] [CrossRef]
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.C.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J. Investig. Dermatol. 2014, 134, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Lee, S.H.; Dolman, C.; Markowitz, D.; Wu, W.; Liu, C.; Reisfeld, R.A.; et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Investig. 2006, 116, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Gray-Owen, S.D.; Blumberg, R.S. CEACAM1: Contact-dependent control of immunity. Nat. Rev. Immunol. 2006, 6, 433–446. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Hanks, B.A.; Holtzhausen, A.; Evans, K.; Heid, M.; Blobe, G.C. Combinatorial TGF-β signaling blockade and anti-CTLA-4 antibody immunotherapy in a murine BRAFV600E-PTEN-/- transgenic model of melanoma. J. Clin. Oncol. 2014, 32, 3011. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Chang, D.K.; Sui, J.; Geng, S.; Muvaffak, A.; Bai, M.; Fuhlbrigge, R.C.; Lo, A.; Yammanuru, A.; Hubbard, L.; Sheehan, J.; et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol. Cancer Ther. 2012, 11, 2451–2461. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Servais, C.; Erez, N. From sentinel cells to inflammatory culprits: Cancer-associated fibroblasts in tumour-related inflammation. J. Pathol. 2013, 229, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Pala, L.; Bagnardi, V.; De Pas, T.; Martinetti, M.; Viale, G.; Gelber, R.D.; Goldhirsch, A. Cancer immunotherapy efficacy and patients’ sex: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 737–746. [Google Scholar] [CrossRef]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.A.; Ganesan, A.P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef]
- Nishijima, T.F.; Muss, H.B.; Shachar, S.S.; Moschos, S.J. Comparison of efficacy of immune checkpoint inhibitors (ICIs) between younger and older patients: A systematic review and meta-analysis. Cancer Treat. Rev. 2016, 45, 30–37. [Google Scholar] [CrossRef]
- Murphy, W.J.; Longo, D.L. The Surprisingly Positive Association Between Obesity and Cancer Immunotherapy Efficacy. JAMA 2019, 321, 1247–1248. [Google Scholar] [CrossRef]
- Jacobi, O.; Landman, Y.; Reinhorn, D.; Icht, O.; Sternschuss, M.; Rotem, O.; Finkel, I.; Allen, A.M.; Dudnik, E.; Goldstein, D.A.; et al. The Relationship of Diabetes Mellitus to Efficacy of Immune Checkpoint Inhibitors in Patients with Advanced Non-Small Cell Lung Cancer. Oncology 2021, 99, 555–561. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Zhou, C.B.; Zhou, Y.L.; Fang, J.Y. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 2021, 7, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Griffin Matthew, E.; Espinosa, J.; Becker Jessica, L.; Luo, J.-D.; Carroll Thomas, S.; Jha Jyoti, K.; Fanger Gary, R.; Hang Howard, C. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 2021, 373, 1040–1046. [Google Scholar] [CrossRef]
- Tray, N.; Weber, J.S.; Adams, S. Predictive Biomarkers for Checkpoint Immunotherapy: Current Status and Challenges for Clinical Application. Cancer Immunol. Res. 2018, 6, 1122–1128. [Google Scholar] [CrossRef]
- Roszik, J.; Haydu, L.E.; Hess, K.R.; Oba, J.; Joon, A.Y.; Siroy, A.E.; Karpinets, T.V.; Stingo, F.C.; Baladandayuthapani, V.; Tetzlaff, M.T.; et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016, 14, 168. [Google Scholar] [CrossRef]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Edenfield, W.J.; Chung, K.; O’Rourke, M.; Cull, E.; Martin, J.; Bowers, H.; Smith, W.; Gluck, W.L. A Phase II Study of Durvalumab in Combination with Tremelimumab in Patients with Rare Cancers. Oncologist 2021, 26, e1499–e1507. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.J.; Eroglu, Z.; Brockstein, B.; Poklepovic, A.S.; Bajaj, M.; Babu, S.; Hallmeyer, S.; Velasco, M.; Lutzky, J.; Higgs, E.; et al. Pembrolizumab Plus Ipilimumab Following Anti-PD-1/L1 Failure in Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 2647–2655. [Google Scholar] [CrossRef]
- Friedman, C.F.; Spencer, C.; Cabanski, C.R.; Panageas, K.S.; Wells, D.K.; Ribas, A.; Tawbi, H.; Tsai, K.; Postow, M.; Shoushtari, A.; et al. Ipilimumab alone or in combination with nivolumab in patients with advanced melanoma who have progressed or relapsed on PD-1 blockade: Clinical outcomes and translational biomarker analyses. J. Immunother. Cancer 2022, 10, e003853. [Google Scholar] [CrossRef]
- Gibney, G.T.; Hamid, O.; Lutzky, J.; Olszanski, A.J.; Mitchell, T.C.; Gajewski, T.F.; Chmielowski, B.; Hanks, B.A.; Zhao, Y.; Newton, R.C.; et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J. Immunother. Cancer 2019, 7, 80. [Google Scholar] [CrossRef]
- Zakharia, Y.; McWilliams, R.R.; Rixe, O.; Drabick, J.; Shaheen, M.F.; Grossmann, K.F.; Kolhe, R.; Pacholczyk, R.; Sadek, R.; Tennant, L.L.; et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother. Cancer 2021, 9, e002057. [Google Scholar] [CrossRef]
- Hawkes, E.A.; Phillips, T.; Budde, L.E.; Santoro, A.; Saba, N.S.; Roncolato, F.; Gregory, G.P.; Verhoef, G.; Offner, F.; Quero, C.; et al. Avelumab in Combination Regimens for Relapsed/Refractory DLBCL: Results from the Phase Ib JAVELIN DLBCL Study. Target. Oncol. 2021, 16, 761–771. [Google Scholar] [CrossRef]
- Burris, H.A.; Callahan, M.K.; Tolcher, A.W.; Kummar, S.; Falchook, G.S.; Pachynski, R.K.; Tykodi, S.S.; Gibney, G.T.; Seiwert, T.Y.; Gainor, J.F.; et al. Phase 1 safety of ICOS agonist antibody JTX-2011 alone and with nivolumab (nivo) in advanced solid tumors; predicted vs observed pharmacokinetics (PK) in ICONIC. J. Clin. Oncol. 2017, 35, 3033. [Google Scholar] [CrossRef]
- Felip, E.; Lim, F.L.; Johnson, M.; O’Brien, M.; Barlesi, F.; Mazieres, J.; Solomon, B.; Moreno, V.; Boni, V.; Swalduz, A.; et al. 1315P Phase Ib/II open-label, randomised evaluation of atezolizumab (atezo) + CPI-444 vs docetaxel as second/third-line therapy in MORPHEUS-NSCLC (non-small cell lung cancer). Ann. Oncol. 2020, 31, S850. [Google Scholar] [CrossRef]
- Sanborn, R.E.; Pishvaian, M.J.; Callahan, M.K.; Weise, A.; Sikic, B.I.; Rahma, O.; Cho, D.C.; Rizvi, N.A.; Sznol, M.; Lutzky, J.; et al. Safety, tolerability and efficacy of agonist anti-CD27 antibody (varlilumab) administered in combination with anti-PD-1 (nivolumab) in advanced solid tumors. J. Immunother. Cancer 2022, 10, e005147. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Tannir, N.M.; Bentebibel, S.E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- O’Day, S.; Borges, V.; Chmielowski, B.; Rao, R.; Abu-Khalaf, M.; Stopeck, A.; Lowe, J.; Mattson, P.; Breuer, K.; Gargano, M.; et al. Abstract P2-09-08: Imprime PGG, a novel innate immune modulator, combined with pembrolizumab in a phase 2 multicenter, open label study in chemotherapy-resistant metastatic triple negative breast cancer (TNBC). Cancer Res. 2019, 79, P2-09-08. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef]
- Cassier, P.A.; Garin, G.; Eberst, L.; Delord, J.-P.; Chabaud, S.; Terret, C.; Montane, L.; Bidaux, A.-S.; Laurent, S.; Jaubert, L.; et al. MEDIPLEX: A phase 1 study of durvalumab (D) combined with pexidartinib (P) in patients (pts) with advanced pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC). J. Clin. Oncol. 2019, 37, 2579. [Google Scholar] [CrossRef]
- Lin, C.-C.; Gil-Martin, M.; Bauer, T.M.; Naing, A.; Lim, D.W.-T.; Sarantopoulos, J.; Geva, R.; Ando, Y.; Fan, L.; Choudhury, S.; et al. Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2020, 80, CT171. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab Versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Wong, D.J.L.; Panwar, A.; Rosenberg, A.; Karivedu, V.; Laux, D.E.; Zandberg, D.P.; Bobilev, D.; Zhao, L.; Wooldridge, J.; Krieg, A.M.; et al. CMP-001-007: Open-label, phase 2 study of intratumoral CMP-001 + pembrolizumab in patients with recurrent or metastatic head and neck squamous cell carcinoma. J. Clin. Oncol. 2021, 39, TPS6089. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef]
- Zamarin, D.; Walderich, S.; Holland, A.; Zhou, Q.; Iasonos, A.E.; Torrisi, J.M.; Merghoub, T.; Chesebrough, L.F.; McDonnell, A.S.; Gallagher, J.M.; et al. Safety, immunogenicity, and clinical efficacy of durvalumab in combination with folate receptor alpha vaccine TPIV200 in patients with advanced ovarian cancer: A phase II trial. J. Immunother. Cancer 2020, 8, e000829. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Gray, J.E.; Devarakonda, S.; Gurtler, J.; Birhiray, R.; Paschold, E.; Dasgupta, A.; Heery, C.; Pico-Navarro, C.; Piechatzek, M.; et al. 1207P—Phase I trial of CV301 in combination with anti-PD-1 therapy in non-squamous NSCLC. Ann. Oncol. 2019, 30, v493–v494. [Google Scholar] [CrossRef]
- Xu, R.-h.; Mai, H.-Q.; Chen, Q.-Y.; Chen, D.; Hu, C.; Yang, K.; Wen, J.; Li, J.-G.; Shi, Y.; Jin, F.; et al. JUPITER-02: Randomized, double-blind, phase III study of toripalimab or placebo plus gemcitabine and cisplatin as first-line treatment for recurrent or metastatic nasopharyngeal carcinoma (NPC). J. Clin. Oncol. 2021, 39, LBA2. [Google Scholar] [CrossRef]
- Locke, F.L.; Westin, J.R.; Miklos, D.B.; Herrara, A.F.; Jacobson, C.A.; Lee, J.; Rossi, J.M.; Bot, A.; Xue, A.; Navale, L.; et al. Phase 1 Results from ZUMA-6: Axicabtagene Ciloleucel (axi-cel; KTE-C19) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma (DLBCL). Blood 2017, 130, 2826. [Google Scholar] [CrossRef]
- Chesney, J.A.; Lutzky, J.; Thomas, S.S.; Nieva, J.J.; Munoz Couselo, E.; Martin-Liberal, J.; Rodriguez-Moreno, J.F.; Cacovean, A.; Li, H.; Fardis, M.; et al. A phase II study of autologous tumor infiltrating lymphocytes (TIL, LN-144/LN-145) in patients with solid tumors. J. Clin. Oncol. 2019, 37, TPS2648. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ribas, A.; Algazi, A.; Ascierto, P.A.; Butler, M.O.; Chandra, S.; Gordon, M.; Hernandez-Aya, L.; Lawrence, D.; Lutzky, J.; Miller, W.H.; et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat. Commun. 2020, 11, 6262. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef]
- Hassel, J.C.; Lee, S.B.; Meiss, F.; Meier, F.; Dimitrakopoulou-Strauss, A.; Jäger, D.; Enk, A.H. Vemurafenib and ipilimumab: A promising combination? Results of a case series. Oncoimmunology 2016, 5, e1101207. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Plimack, E.R.; Ernstoff, M.S.; Lewis, L.D.; Bauer, T.M.; McDermott, D.F.; Carducci, M.; Kollmannsberger, C.; Rini, B.I.; Heng, D.Y.C.; et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: The CheckMate 016 study. J. Immunother. Cancer 2018, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Kelly, K.; Daly, M.E.; Mirhadi, A.; Lara, F.; Garcia, L.A.; Chen, S.; Eastham, D.; Wiegner, E.A.; Riess, J.W.; Schalper, K.A.; et al. Atezolizumab plus stereotactic ablative therapy for medically inoperable patients with early-stage non-small cell lung cancer. J. Clin. Oncol. 2020, 38, 9011. [Google Scholar] [CrossRef]
- Theelen, W.; Peulen, H.M.U.; Lalezari, F.; van der Noort, V.; de Vries, J.F.; Aerts, J.; Dumoulin, D.W.; Bahce, I.; Niemeijer, A.N.; de Langen, A.J.; et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients with Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1276–1282. [Google Scholar] [CrossRef]
- Maity, A.; Mick, R.; Huang, A.C.; George, S.M.; Farwell, M.D.; Lukens, J.N.; Berman, A.T.; Mitchell, T.C.; Bauml, J.; Schuchter, L.M.; et al. A phase I trial of pembrolizumab with hypofractionated radiotherapy in patients with metastatic solid tumours. Br. J. Cancer 2018, 119, 1200–1207. [Google Scholar] [CrossRef]
- Foster, C.C.; Fleming, G.F.; Karrison, T.G.; Liao, C.Y.; Desai, A.V.; Moroney, J.W.; Ratain, M.J.; Nanda, R.; Polite, B.N.; Hahn, O.M.; et al. Phase I Study of Stereotactic Body Radiotherapy plus Nivolumab and Urelumab or Cabiralizumab in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5510–5518. [Google Scholar] [CrossRef]
- Jotte, R.; Cappuzzo, F.; Vynnychenko, I.; Stroyakovskiy, D.; Rodríguez-Abreu, D.; Hussein, M.; Soo, R.; Conter, H.J.; Kozuki, T.; Huang, K.C.; et al. Atezolizumab in Combination with Carboplatin and Nab-Paclitaxel in Advanced Squamous NSCLC (IMpower131): Results from a Randomized Phase III Trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1351–1360. [Google Scholar] [CrossRef]
- Cortés, J.; André, F.; Gonçalves, A.; Kümmel, S.; Martín, M.; Schmid, P.; Schuetz, F.; Swain, S.M.; Easton, V.; Pollex, E.; et al. IMpassion132 Phase III trial: Atezolizumab and chemotherapy in early relapsing metastatic triple-negative breast cancer. Future Oncol. 2019, 15, 1951–1961. [Google Scholar] [CrossRef]
- Pusztai, L.; Yau, C.; Wolf, D.M.; Han, H.S.; Du, L.; Wallace, A.M.; String-Reasor, E.; Boughey, J.C.; Chien, A.J.; Elias, A.D.; et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 2021, 39, 989–998.e985. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in Combination with Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Carter, C.; Caroen, S.; Oronsky, B.; Quinn, M.F.; Williams, J.; Brzezniak, C.E. Phase I pilot study of RRx-001 + nivolumab in patients with traditionally non-checkpoint inhibitor-responsive cancers (PRIMETIME). J. Clin. Oncol. 2020, 38, e15119. [Google Scholar] [CrossRef]
- Iyer, S.P.; Xu, J.; Becnel, M.R.; Nair, R.; Steiner, R.; Feng, L.; Lee, H.J.; Strati, P.; Ahmed, S.; Parmar, S.; et al. A Phase II Study of Pembrolizumab in Combination with Romidepsin Demonstrates Durable Responses in Relapsed or Refractory T-Cell Lymphoma (TCL). Blood 2020, 136, 40–41. [Google Scholar] [CrossRef]
- Segal, N.H.; Infante, J.R.; Sanborn, R.E.; Gibney, G.T.; Lawrence, D.P.; Rizvi, N.; Leidner, R.; Gajewski, T.F.; Bertino, E.; Sharfman, W.H.; et al. 1086P—Safety of the natural killer (NK) cell-targeted anti-KIR antibody, lirilumab (liri), in combination with nivolumab (nivo) or ipilimumab (ipi) in two phase 1 studies in advanced refractory solid tumors. Ann. Oncol. 2016, 27, vi372. [Google Scholar] [CrossRef]
- Hanna, G.J.; O’Neill, A.; Shin, K.Y.; Wong, K.; Jo, V.Y.; Quinn, C.T.; Cutler, J.M.; Flynn, M.; Lizotte, P.H.; Annino, D.J., Jr.; et al. Neoadjuvant and Adjuvant Nivolumab and Lirilumab in Patients with Recurrent, Resectable Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 468–478. [Google Scholar] [CrossRef]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef]
- Silk, A.W.; O’Day, S.J.; Kaufman, H.L.; Bryan, J.; Norrell, J.T.; Imbergamo, C.; Portal, D.; Zambrano-Acosta, E.; Palmeri, M.; Fein, S.; et al. Abstract CT139: Intratumoral oncolytic virus V937 in combination with pembrolizumab (pembro) in patients (pts) with advanced melanoma: Updated results from the phase 1b CAPRA study. Cancer Res. 2021, 81, CT139. [Google Scholar] [CrossRef]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, Y.; Li, Q.; Yao, J.; Yuan, X.; Zhang, Y.; Yin, X.; Saito, Y.; Fan, H.; Li, P.; et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients via activation of the macrophage histamine receptor H1. Cancer Cell 2022, 40, 36–52.e39. [Google Scholar] [CrossRef]
- Chou, C.; Zhang, X.; Krishna, C.; Nixon, B.G.; Dadi, S.; Capistrano, K.J.; Kansler, E.R.; Steele, M.; Han, J.; Shyu, A.; et al. Programme of self-reactive innate-like T cell-mediated cancer immunity. Nature 2022, 605, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e914. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Gupta, J.; Safdari, H.A.; Hoque, M. Nanoparticle mediated cancer immunotherapy. Semin. Cancer Biol. 2021, 69, 307–324. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, T.; Endo, R.; Sasaki, S.; Takahashi, N.; Sato, Y.; Hyodo, M.; Hayakawa, Y.; Harashima, H. STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J. Immunother. Cancer 2021, 9, e002852. [Google Scholar] [CrossRef]
- Li, X.; Khorsandi, S.; Wang, Y.; Santelli, J.; Huntoon, K.; Nguyen, N.; Yang, M.; Lee, D.; Lu, Y.; Gao, R.; et al. Cancer immunotherapy based on image-guided STING activation by nucleotide nanocomplex-decorated ultrasound microbubbles. Nat. Nanotechnol. 2022, 30, 1–9. [Google Scholar] [CrossRef]
- Buss, C.G.; Bhatia, S.N. Nanoparticle delivery of immunostimulatory oligonucleotides enhances response to checkpoint inhibitor therapeutics. Proc. Natl. Acad. Sci. USA 2020, 117, 13428–13436. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xu, L.; Chao, Y.; Xu, J.; Sun, X.; Wu, Y.; Peng, R.; Liu, Z. Hollow MnO(2) as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat. Commun. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- de Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad. Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Silva, A.L.; Camps, M.; Allam, A.; Jiskoot, W.; van der Burg, S.H.; Ossendorp, F.; Oostendorp, J. Efficient ex vivo induction of T cells with potent anti-tumor activity by protein antigen encapsulated in nanoparticles. Cancer Immunol. Immunother. 2013, 62, 1161–1173. [Google Scholar] [CrossRef]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | Approach | References |
|---|---|---|
| 1. | Combination checkpoint blockade | [124,125,126] |
| 2. | Checkpoint blockade plus metabolic modulators | [127,128] |
| 3. | Checkpoint blockade plus other immune modulators | [129,130,131,132,133,134,135] |
| 4. | Checkpoint blockade plus macrophage inhibitors | [136,137] |
| 5. | Checkpoint blockade plus oncolytic viruses or plus TLR agonists | [138,139,140] |
| 6. | Checkpoint blockade plus cancer vaccines | [141,142,143,144] |
| 7. | Checkpoint blockade plus ACT | [145,146] |
| 8. | Checkpoint blockade plus targeted therapies | [147,148,149,150,151,152,153,154] |
| 9. | Checkpoint blockade plus radiation therapy | [155,156,157,158] |
| 10. | Checkpoint blockade plus chemotherapy | [159,160,161,162,163] |
| 11. | Checkpoint blockade plus epigenetic modifications | [164,165] |
| 12. | Checkpoint blockade plus NK activation | [166,167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, S.H.; Vetrivel, P.; Kim, J.; Lee, M.-S. Cancer Resistance to Immunotherapy: Molecular Mechanisms and Tackling Strategies. Int. J. Mol. Sci. 2022, 23, 10906. https://doi.org/10.3390/ijms231810906
Vu SH, Vetrivel P, Kim J, Lee M-S. Cancer Resistance to Immunotherapy: Molecular Mechanisms and Tackling Strategies. International Journal of Molecular Sciences. 2022; 23(18):10906. https://doi.org/10.3390/ijms231810906
Chicago/Turabian StyleVu, Son Hai, Preethi Vetrivel, Jongmin Kim, and Myeong-Sok Lee. 2022. "Cancer Resistance to Immunotherapy: Molecular Mechanisms and Tackling Strategies" International Journal of Molecular Sciences 23, no. 18: 10906. https://doi.org/10.3390/ijms231810906
APA StyleVu, S. H., Vetrivel, P., Kim, J., & Lee, M.-S. (2022). Cancer Resistance to Immunotherapy: Molecular Mechanisms and Tackling Strategies. International Journal of Molecular Sciences, 23(18), 10906. https://doi.org/10.3390/ijms231810906