
LSSmScarlet2 and LSSmScarlet3, Chemically Stable Genetically Encoded Red Fluorescent Proteins with a Large Stokes’ Shift
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
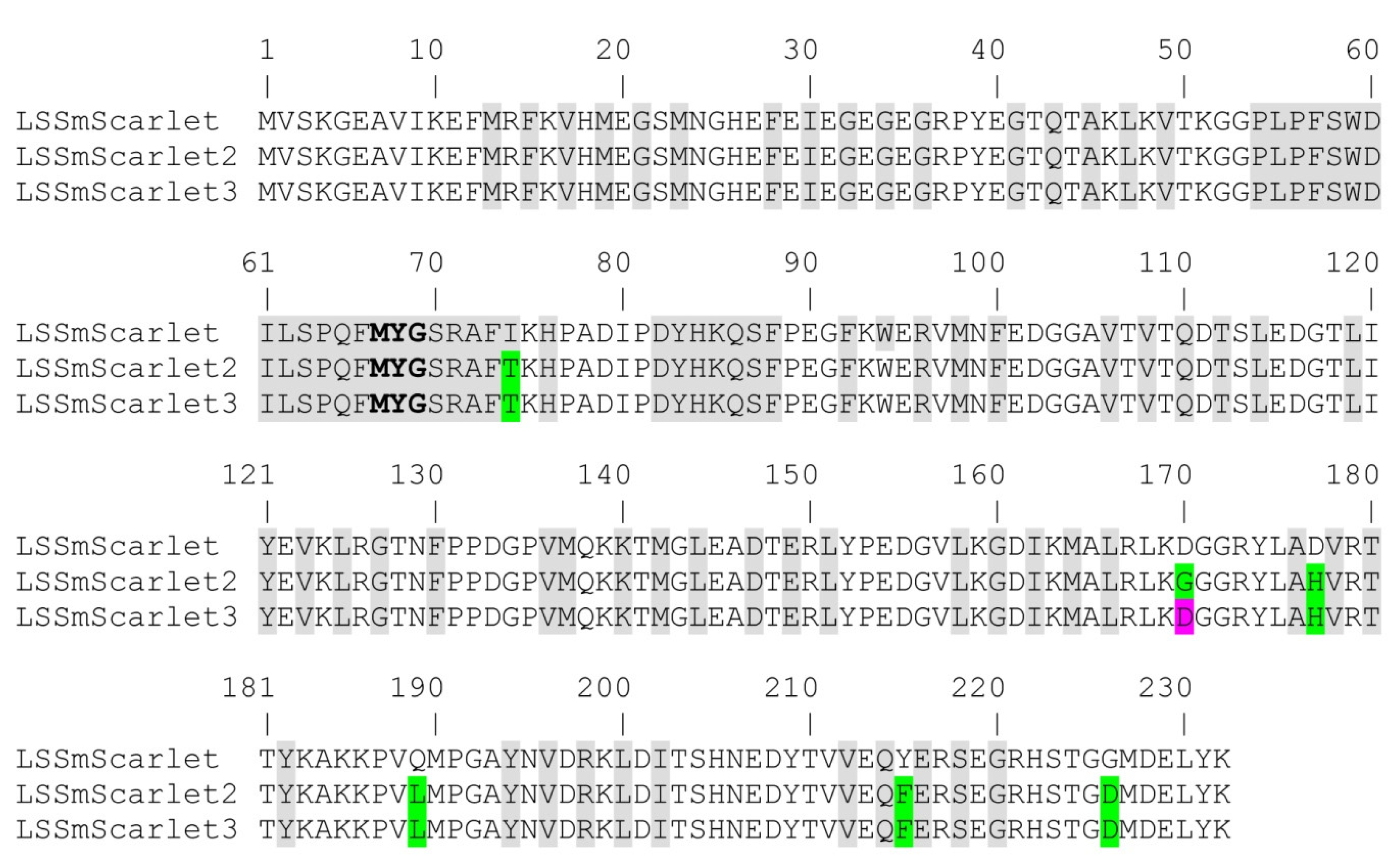
2.1. Developing the Large Stokes’ Shift Fluorescent Protein LSSmScarlet2 in E. coli
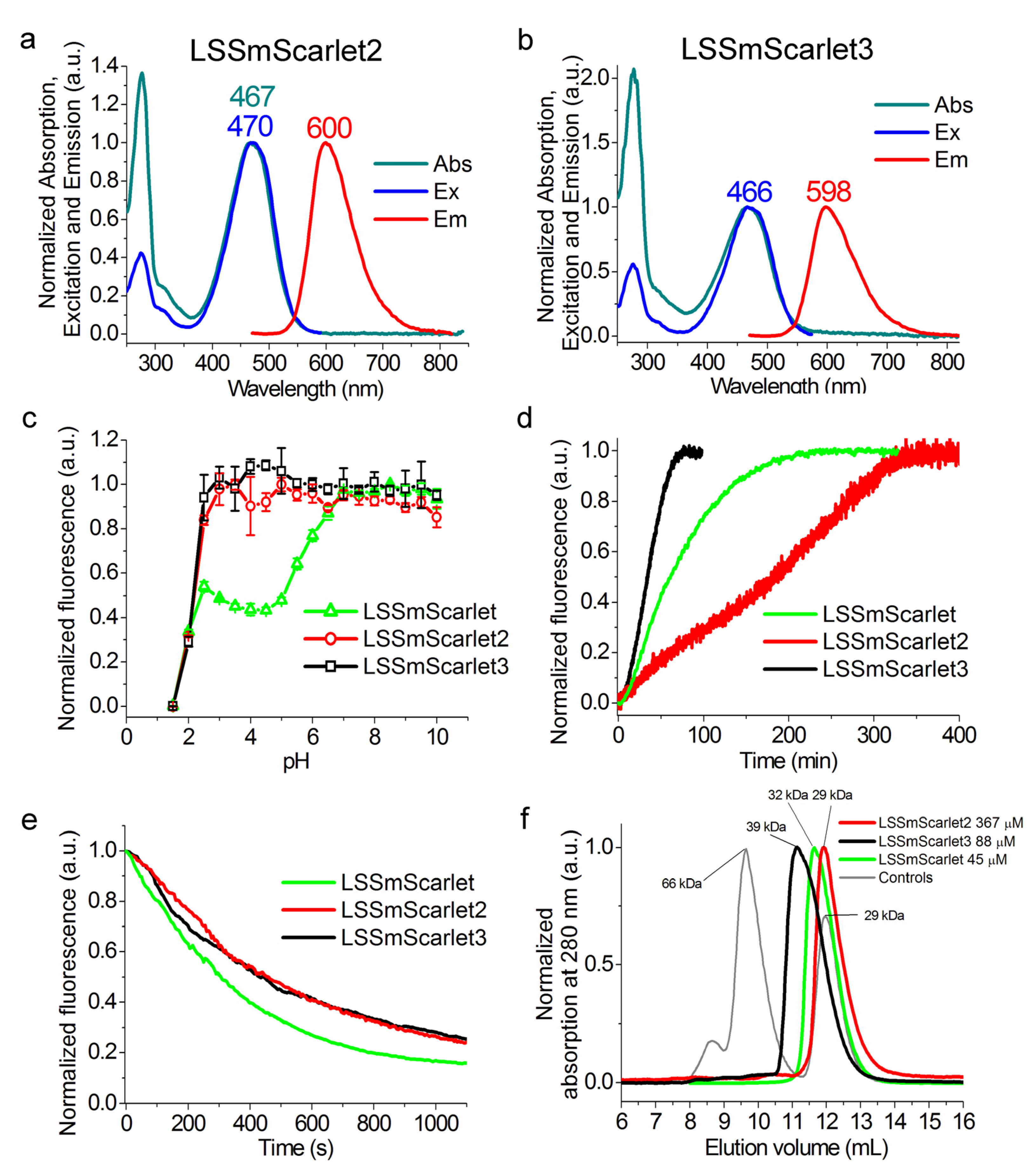
2.2. In Vitro Characterization of the Purified LSSmScarlet2 Protein
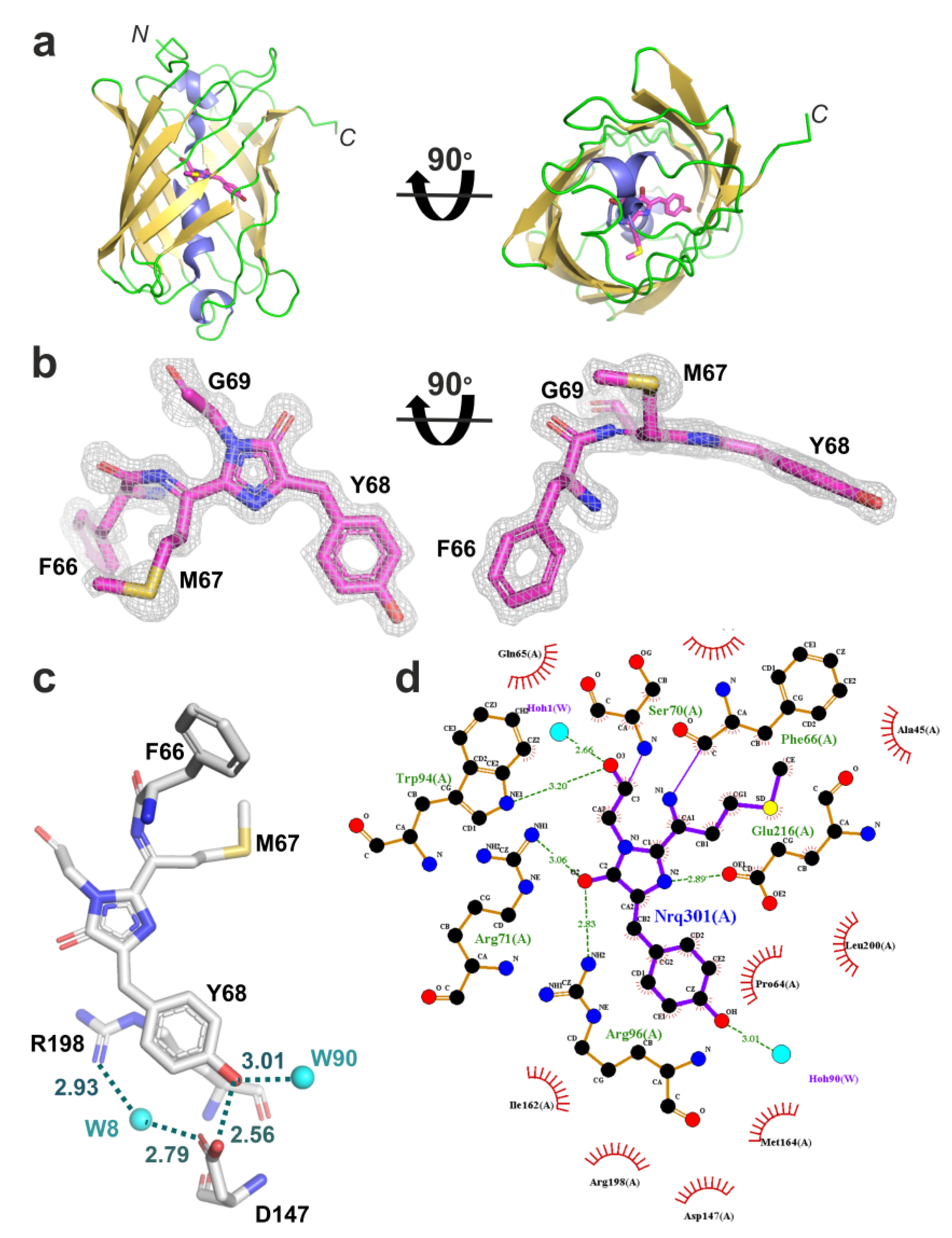
2.3. Structural Characterization of the LSSmScarlet2 Protein
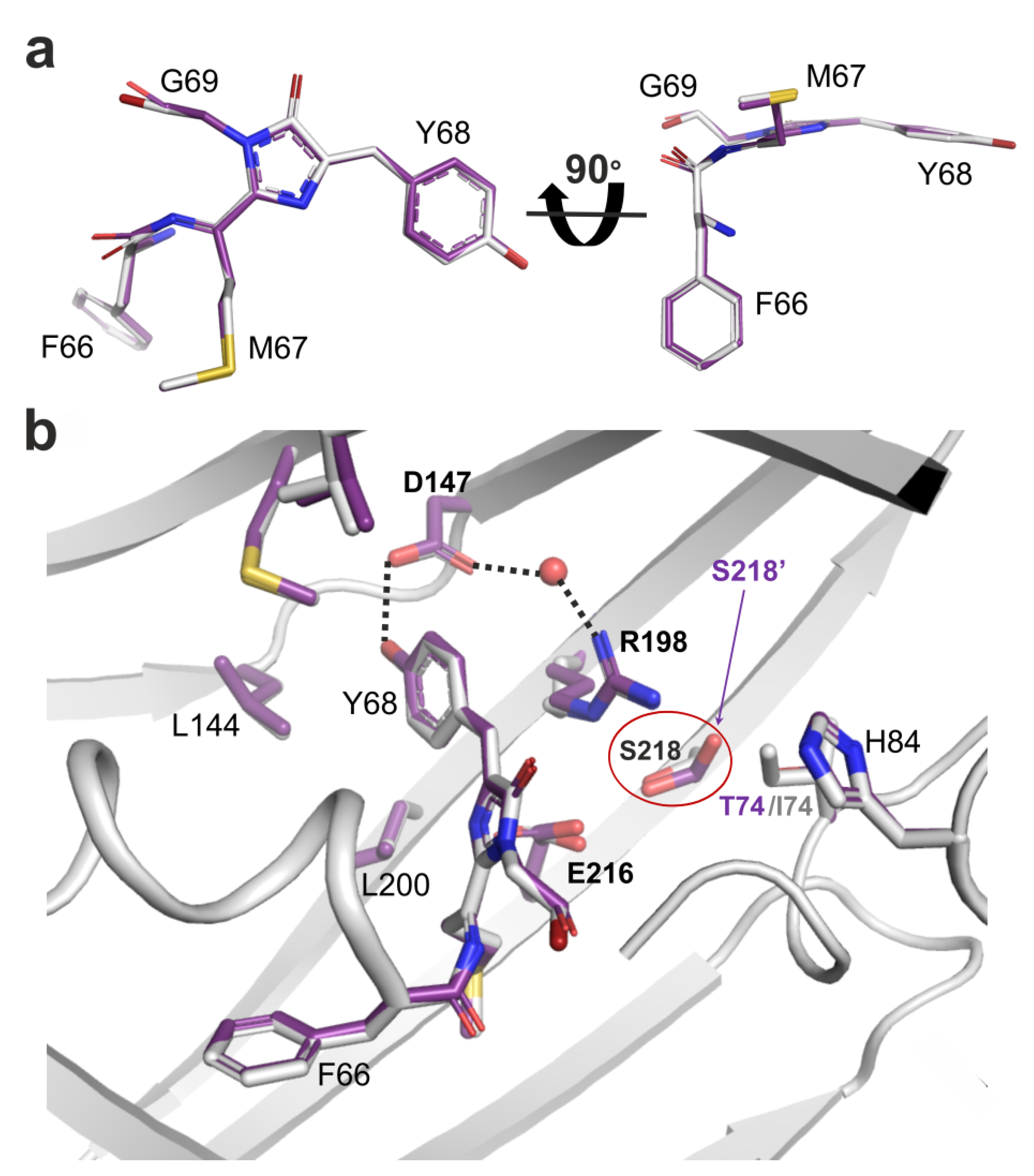
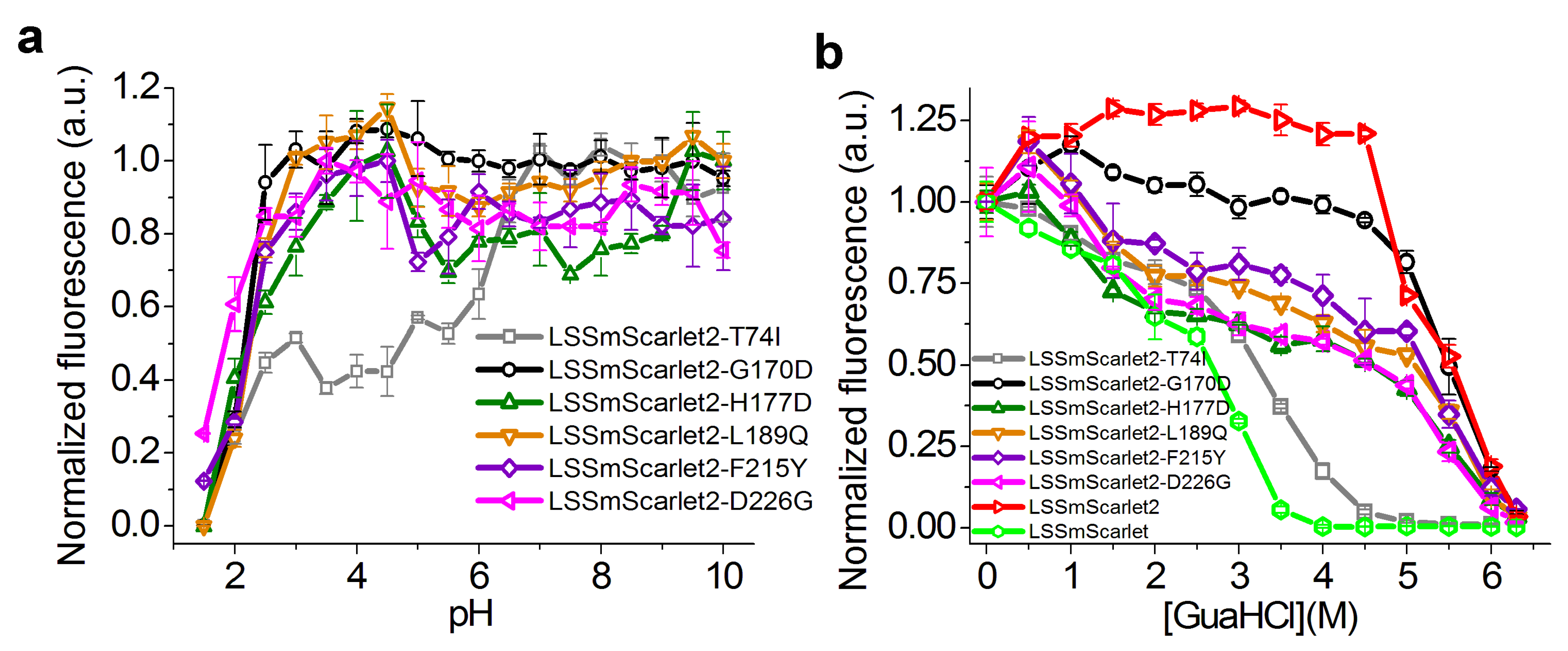
2.4. Site-Directed Mutagenesis of the LSSmScarlet2 LSSRFP
2.5. In Vitro Characterization of the Purified LSSmScarlet3 Protein
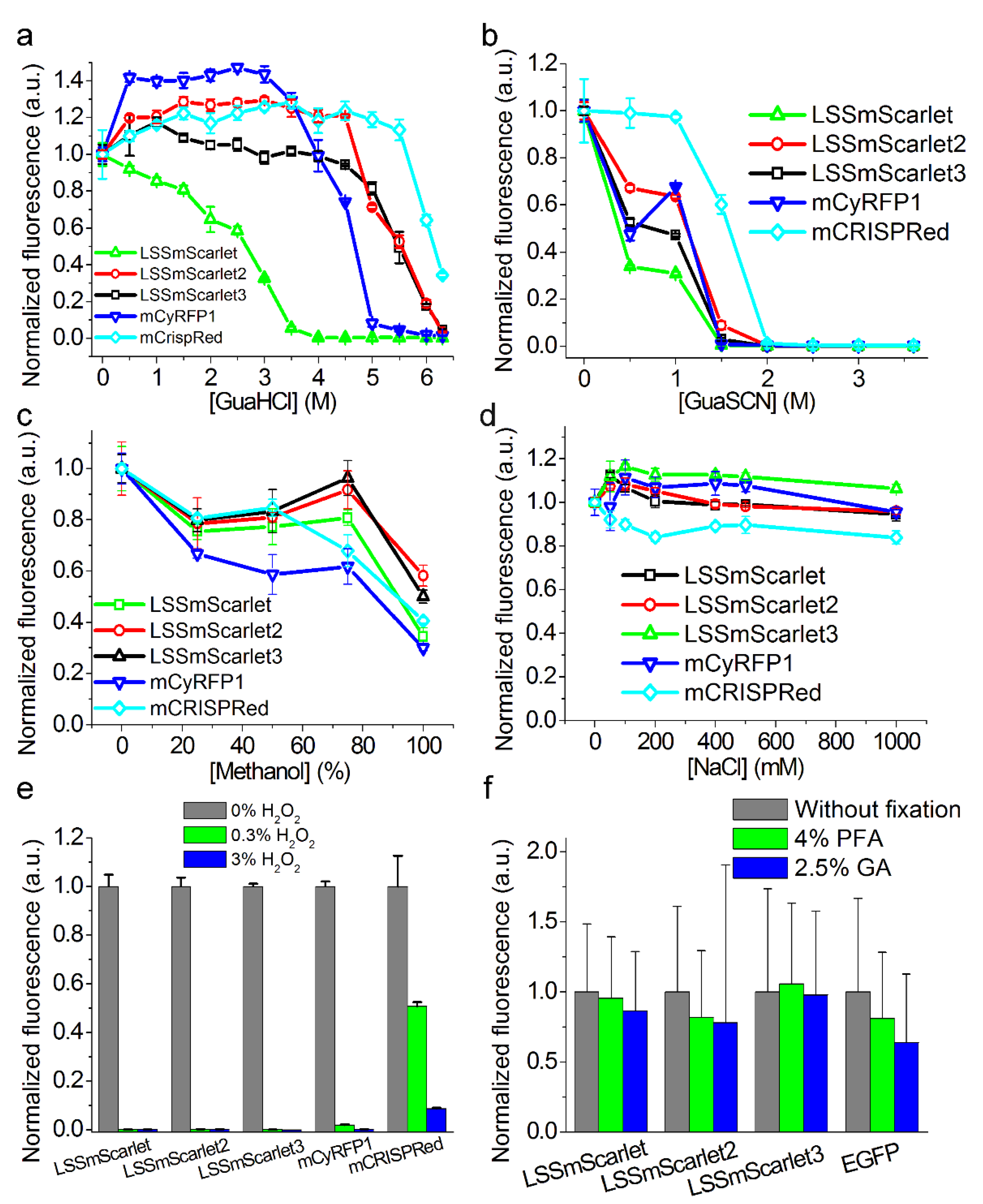
2.6. Chemical Stability of LSSmScarlet2 and LSSmScarlet3 LSSRFPs
2.7. Brightness of LSSmScarlet2 and LSSmScarlet3 in the Cytosol of Cultured Mammalian Cells
2.8. Behavior of LSSmScarlet2 and LSSmScarlet3 in Mammalian Cells in Fusions with Structural and Mitochondrial Proteins
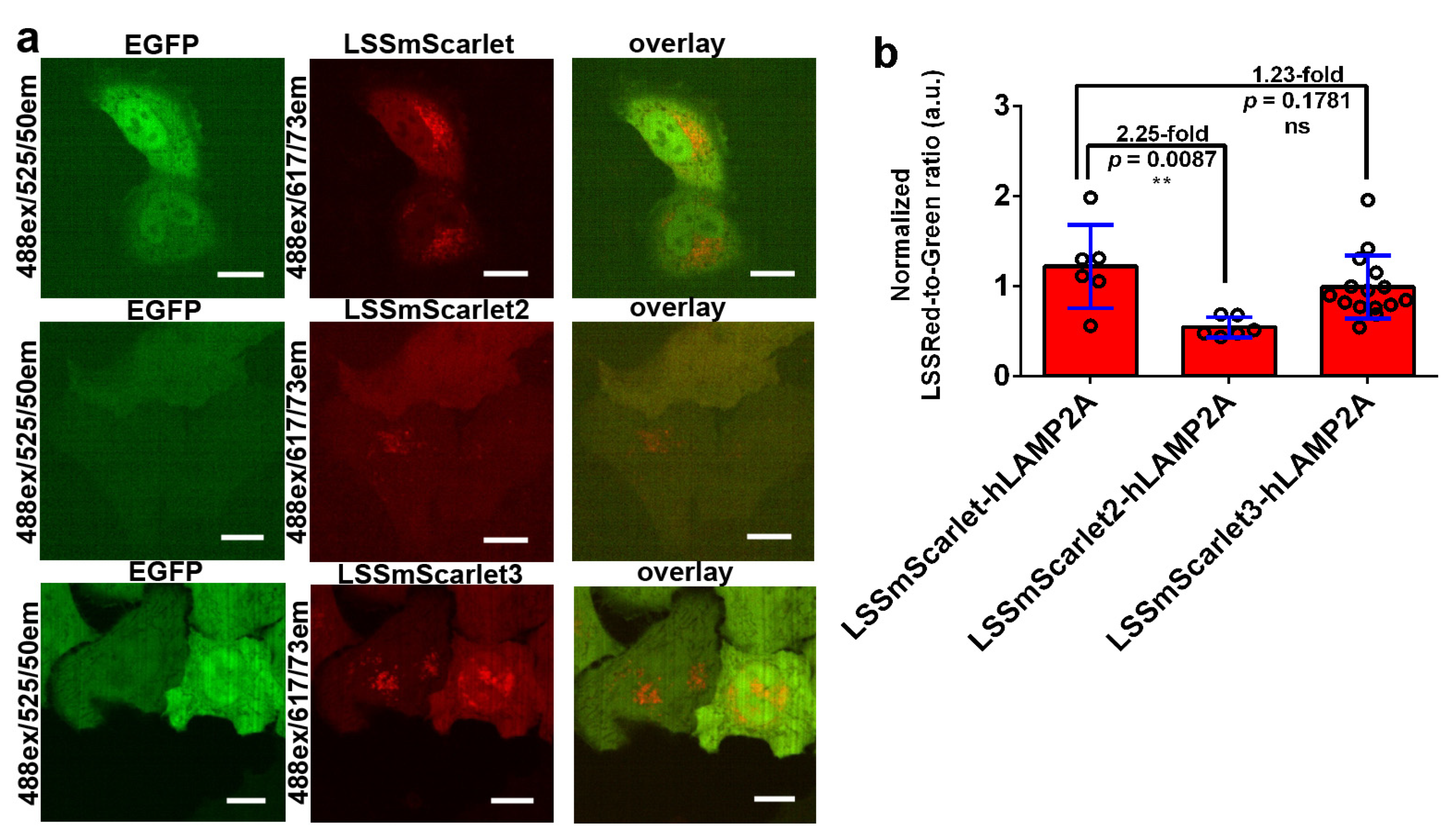
2.9. Brightness of LSSmScarlet2 and LSSmScarlet3 in Lysosomes of Cultured Mammalian Cells
3. Materials and Methods
3.1. Cloning of Bacterial Vectors, Mutagenesis, and Library Screening
3.2. Protein Purification and Characterization
3.3. Protein Crystallization
3.4. Data Collection, Processing, Structure Solution, and Refinement
3.5. Structure Analysis and Validation
3.6. Chemical Stability
3.7. Mammalian Plasmid Construction
3.8. Mammalian Live- and Fixed-Cell Imaging
3.9. Cell Fixation with 4% PFA and 2.5% GA
3.10. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FP | Fluorescent protein |
| RFP | Red fluorescent protein |
| LSSRFP | Red fluorescent protein with a large Stokes’ shift |
| PBS | Phosphate-buffered saline |
| QY | Quantum yield |
| SD | Standard deviation |
References
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Oh, Y.; Sens, A.; Ataie, N.; Dana, H.; Macklin, J.J.; Laviv, T.; Welf, E.S.; Dean, K.M.; Zhang, F.; et al. A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol. 2016, 34, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, K.; Nienhaus, G.U. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2014, 43, 1088–1106. [Google Scholar] [CrossRef] [PubMed]
- Tillberg, P.W.; Chen, F.; Piatkevich, K.D.; Zhao, Y.; Yu, C.C.; English, B.P.; Gao, L.; Martorell, A.; Suk, H.J.; Yoshida, F.; et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 2016, 34, 987–992. [Google Scholar] [CrossRef]
- Paez-Segala, M.G.; Sun, M.G.; Shtengel, G.; Viswanathan, S.; Baird, M.A.; Macklin, J.J.; Patel, R.; Allen, J.R.; Howe, E.S.; Piszczek, G.; et al. Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat. Methods 2015, 12, 215–218, 4 p following 218. [Google Scholar] [CrossRef]
- Campbell, B.C.; Nabel, E.M.; Murdock, M.H.; Lao-Peregrin, C.; Tsoulfas, P.; Blackmore, M.G.; Lee, F.S.; Liston, C.; Morishita, H.; Petsko, G.A. mGreenLantern: A bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. Proc. Natl. Acad. Sci. USA 2020, 117, 30710–30721. [Google Scholar] [CrossRef]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS ONE 2011, 6, e28674. [Google Scholar] [CrossRef]
- Erdogan, M.; Fabritius, A.; Basquin, J.; Griesbeck, O. Targeted In Situ Protein Diversification and Intra-organelle Validation in Mammalian Cells. Cell Chem. Biol. 2020, 27, 610–621.e5. [Google Scholar] [CrossRef]
- Subach, O.M.; Vlaskina, A.V.; Agapova, Y.K.; Dorovatovskii, P.V.; Nikolaeva, A.Y.; Ivashkina, O.I.; Popov, V.O.; Piatkevich, K.D.; Khrenova, M.G.; Smirnova, T.A.; et al. LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift. Int. J. Mol. Sci. 2021, 22, 12887. [Google Scholar] [CrossRef]
- Proksch, E. pH in nature, humans and skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Laviv, T.; Kim, B.B.; Chu, J.; Lam, A.J.; Lin, M.Z.; Yasuda, R. Simultaneous dual-color fluorescence lifetime imaging with novel red-shifted fluorescent proteins. Nat. Methods 2016, 13, 989–992. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Kuznetsova, I.M.; Verkhusha, V.V.; Turoverov, K.K. Sensitivity of superfolder GFP to ionic agents. PLoS ONE 2014, 9, e110750. [Google Scholar] [CrossRef]
- Lazutkin, A.A.; Shuvaev, S.A.; Barykina, N.V. Click histochemistry for whole-mount staining of brain structures. MethodsX 2019, 6, 1986–1991. [Google Scholar] [CrossRef]
- Wachter, R.M.; Remington, S.J. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr. Biol. CB 1999, 9, R628–R629. [Google Scholar] [CrossRef]
- Radulescu, R.T.; Boenisch, T. Blocking endogenous peroxidases: A cautionary note for immunohistochemistry. J. Cell. Mol. Med. 2007, 11, 1419. [Google Scholar]
- Qin, Y.; Jiang, W.; Li, A.; Gao, M.; Liu, H.; Gao, Y.; Tian, X.; Gong, G. The Combination of Paraformaldehyde and Glutaraldehyde Is a Potential Fixative for Mitochondria. Biomolecules 2021, 11, 711. [Google Scholar] [CrossRef]
- Martina, J.A.; Raben, N.; Puertollano, R. SnapShot: Lysosomal Storage Diseases. Cell 2020, 180, 602–602.e1. [Google Scholar] [CrossRef]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef]
- Isenman, L.D.; Dice, J.F. Secretion of intact proteins and peptide fragments by lysosomal pathways of protein degradation. J. Biol. Chem. 1989, 264, 21591–21596. [Google Scholar] [CrossRef]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Pang, S.; Chen, D.; Zhang, A.; Qin, X.; Yan, B. Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson’s disease. Neurosci. Lett. 2012, 526, 63–67. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Cuervo, A.M. Dietary lipids and aging compromise chaperone-mediated autophagy by similar mechanisms. Autophagy 2012, 8, 1152–1154. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (Pt 2), 125–132. [Google Scholar] [CrossRef]
- The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50 (Pt 5), 760–763. [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (Pt 12 Pt 1), 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Barykina, N.V.; Anokhin, K.V.; Piatkevich, K.D.; Subach, F.V. Near-Infrared Genetically Encoded Positive Calcium Indicator Based on GAF-FP Bacterial Phytochrome. Int. J. Mol. Sci. 2019, 20, 3488. [Google Scholar] [CrossRef]
- Subach, O.M.; Kunitsyna, T.A.; Mineyeva, O.A.; Lazutkin, A.A.; Bezryadnov, D.V.; Barykina, N.V.; Piatkevich, K.D.; Ermakova, Y.G.; Bilan, D.S.; Belousov, V.V.; et al. Slowly Reducible Genetically Encoded Green Fluorescent Indicator for In Vivo and Ex Vivo Visualization of Hydrogen Peroxide. Int. J. Mol. Sci. 2019, 20, 3138. [Google Scholar] [CrossRef]
- Subach, O.M.; Gundorov, I.S.; Yoshimura, M.; Subach, F.V.; Zhang, J.; Gruenwald, D.; Souslova, E.A.; Chudakov, D.M.; Verkhusha, V.V. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 2008, 15, 1116–1124. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Abs, Ex/Em (nm) | QY (%) a | ε (mM−1·cm−1) b | Brightness vs. EGFP (%) | pKa | Monomeric State | Photobleaching Half-Time (s) e | Maturation Half-Time (min) | |
|---|---|---|---|---|---|---|---|---|---|
| Ex. at Max c | Ex. at 488 nm d | ||||||||
| LSSmScarlet | 466, 470/598 | 43 ± 2 | 30.2 ± 0.6 | 39 | 34 | 1.91 ± 0.01; 5.78 ± 0.06 | Monomer | 310 ± 53 | 61 |
| LSSmScarlet2 | 467, 470/600 | 29 ± 2 | 30.0 ± 1.2 | 26 | 23.5 | 2.19 ± 0.01 | Monomer | 487 ± 148 | 184 |
| LSSmScarlet3 | 466,466/598 | 36 ± 1 | 27.3 ± 1.4 | 29 | 26 | 2.18 ± 0.01 | Monomer | 463 ± 237 | 34 |
| Data Collection | |
|---|---|
| Diffraction Source | ESRF, ID30A |
| Wavelength (Å) | 0.967697 |
| Resolution range (Å) | 39.8–1.41 |
| Detector | Eiger 4M |
| Space group | C2 |
| a, b, c (Å) | 84.62; 45.37; 58.74 |
| α, β, γ (°) | 90.0; 102.09; 90.0 |
| Unique reflections | 42,307 (2725) |
| Resolution range (Å) | 39.8–1.41 (1.45–1.41) * |
| Completeness (%) | 97.57 (91.7) |
| Multiplicity | 1.56 (1.38) |
| 〈I/σ(I)〉 | 15.5 (4.43) |
| Rmeas (%) | 5.8 (25.8) |
| CC1/2 Wilson B-factor (Å2) | 99.8 (93.8) 17.61 |
| Refinement | |
| Resolution range | 15–1.41 |
| Rwork/Rfree (%) | 11.25/15.23 |
| Bonds (Å) | 0.011 |
| Angles (°) | 1.7525 |
| Ramachandran plot | |
| Most favored (%) | 95.5 |
| Allowed (%) | 4.5 |
| No. atoms | |
| Protein | 1888 |
| Water | 381 |
| Chromophore | 23 |
| Other ligands | 5 |
| B-factors (Å2) | |
| Protein | 10.8 |
| Water | 21.34 |
| Chromophore | 9.7 |
| Other ligands | 21.8 |
| Protein | pKa | GuaHCl Concentration at Half-Maximal Fluorescence (M) |
|---|---|---|
| LSSmScarlet | 1.91 ± 0.01; 5.78 ± 0.06 | 2.66 ± 0.10 |
| LSSmScarlet2 | 2.19 ± 0.01 | 5.50 ± 0.05 |
| LSSmScarlet3 | 2.18 ± 0.01 | 5.49 ± 0.13 |
| LSSmScarlet2/T74I | 2.03 ± 0.04 6.16 ± 0.09 | 3.20 ± 0.06 |
| LSSmScarlet2/H177D | 2.22 ± 0.16 | 4.55 ± 0.13 |
| LSSmScarlet2/L189Q | 2.33 ± 0.02 | 4.86 ± 0.44 |
| LSSmScarlet2/F215Y | 2.25 ± 0.01 | 5.25 ± 0.09 |
| LSSmScarlet2/D226G | 1.88 ± 0.06 | 4.61 ± 0.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subach, O.M.; Vlaskina, A.V.; Agapova, Y.K.; Piatkevich, K.D.; Patrushev, M.V.; Samygina, V.R.; Subach, F.V. LSSmScarlet2 and LSSmScarlet3, Chemically Stable Genetically Encoded Red Fluorescent Proteins with a Large Stokes’ Shift. Int. J. Mol. Sci. 2022, 23, 11051. https://doi.org/10.3390/ijms231911051
Subach OM, Vlaskina AV, Agapova YK, Piatkevich KD, Patrushev MV, Samygina VR, Subach FV. LSSmScarlet2 and LSSmScarlet3, Chemically Stable Genetically Encoded Red Fluorescent Proteins with a Large Stokes’ Shift. International Journal of Molecular Sciences. 2022; 23(19):11051. https://doi.org/10.3390/ijms231911051
Chicago/Turabian StyleSubach, Oksana M., Anna V. Vlaskina, Yulia K. Agapova, Kiryl D. Piatkevich, Maxim V. Patrushev, Valeriya R. Samygina, and Fedor V. Subach. 2022. "LSSmScarlet2 and LSSmScarlet3, Chemically Stable Genetically Encoded Red Fluorescent Proteins with a Large Stokes’ Shift" International Journal of Molecular Sciences 23, no. 19: 11051. https://doi.org/10.3390/ijms231911051
APA StyleSubach, O. M., Vlaskina, A. V., Agapova, Y. K., Piatkevich, K. D., Patrushev, M. V., Samygina, V. R., & Subach, F. V. (2022). LSSmScarlet2 and LSSmScarlet3, Chemically Stable Genetically Encoded Red Fluorescent Proteins with a Large Stokes’ Shift. International Journal of Molecular Sciences, 23(19), 11051. https://doi.org/10.3390/ijms231911051