

MPSI Manifestations and Treatment Outcome: Skeletal Focus

 , , and
, , and
Abstract
1. Introduction
2. Pathological Mechanisms Leading to GAG Accumulation
3. Endochondral Bone Formation
3.1. Principal Transcription Factors and Signaling Pathways in Endochondral Bone Formation
- SOX9. The transcription factor SOX9 has a pivotal role in chondrocyte differentiation and cartilage formation [75,76]. In columnar chondrocytes, SOX9 is important to maintain cell proliferation and to directly silence genes that are normally expressed by hypertrophic chondrocytes, such as Col10a1 and Vegfa [77,78]. In contrast, in early pre-hypertrophic chondrocytes, it directly and indirectly activates Col10a1, thus promoting the initial step of hypertrophic differentiation [79]. Finally, SOX9 is downregulated in hypertrophic chondrocytes as a necessary step for vascular invasion and subsequent bone deposition.SOX9 regulates growth plate development and chondrocyte differentiation through multiple mechanisms. For example, SOX9 is able to inhibit WNT signaling, whose members, in particular WNT5A and WNT5B, are known to be important for the progression of chondrocytes to hypertrophic maturation [80]. SOX9 modulates this pathway by interacting directly with βcatenin and promoting its nuclear translocation and degradation by the ubiquitination 26/S proteasome pathway [81,82]. In addition, SOX9 modulates RUNX2, another key transcription factor that directs both chondrocyte maturation and osteogenic differentiation during skeletal development [83]. SOX9 promotes RUNX2 degradation in a proteasome-independent, but phosphorylation-dependent, manner, and the balance between these two transcription factors is critical for correct growth plate formation (Figure 4) [84,85].
- IHH. IHH is essential for embryonic skeletal development, as well to modulate postnatal skeletal growth [86,87]. In the growth plate, IHH is expressed in the hypertrophic zone and regulates both proliferation and differentiation of chondrocytes [86]. Mice with global Ihh knockout show abnormalities in long bones, which are shorter compared to those of wild-type mice due to a marked reduction of chondrocyte proliferation with thinning of the cartilage region. In addition, they display a premature chondrocyte hypertrophy and absence of mature osteoblasts [86]. IHH coordinates events in the endochondral ossification via parathyroid hormone-related protein (PTHrP)-independent or -dependent pathways. In fact, similarly to Ihh−/− mice, transgenic models with the cartilage-specific ablation of Smo, which encodes for a G protein-coupled receptor that transduces the hedgehog’s protein signal, exhibit reduced chondrocyte proliferation, indicating that this process requires a direct IHH input [87]. However, in the same mice, chondrocyte differentiation and maturation appear similarly as in wild-type mice, suggesting that IHH does not regulate directly chondrocyte hypertrophy, but is instead dependent on its action on the PTHrP pathway [87].
- PTHrP. PTHrP is required for the maintenance of chondrocytes in their proliferative phase and to delay the hypertrophic one [88,89]. PTHrP establishes a feedback loop with IHH that is necessary to regulate chondrocyte proliferation and hypertrophy. Specifically, IHH, expressed by hypertrophic chondrocytes, stimulates the chondrocytes close to the articular region to produce PTHrP, which inhibits IHH expression in lower chondrocytes, keeping them proliferating. When chondrocytes are no longer reached by PTHrP, they stop to proliferate and start to secrete IHH, thus closing the loop [90,91,92,93,94].Pthrp−/− mice exhibit a reduction in the height of the proliferative zone, whereas overexpression of Pthrp, specifically in chondrocytes, results in delayed endochondral ossification and chondrodysplasia [95]. Interestingly, it has been shown that the proliferation rate of chondrocytes in Pthrp−/− mice is not affected. This indicates that the depletion of the proliferative zone results from an accelerated rate of chondrocyte differentiation, rather than from a change in the proliferative activity [96]. This is in agreement with the inhibitory function of PTHrP on two transcription factors involved in chondrocyte hypertrophy, such as the myocyte enhancer factor-2 and RUNX2 [97,98]. However, Huang et al. have suggested that PTHrP signaling retains chondrocytes in a proliferative phase also by inducing a PKA-mediated phosphorylation and activation of SOX9 (Figure 4) [99].
- FGFs. The fibroblast growth factor (FGF) protein family consists of 23 members, which have different roles in various biological processes. FGFs bind and activate four receptor tyrosine kinase molecules (FGF receptors, FGFRs), which have a specific and distinct expression pattern in the growth plate. Fgfr1 and Fgfr2 are both expressed in the perichondrium and periosteum; conversely, Fgfr3 is expressed predominately in proliferating chondrocytes and in pre-hypertrophic chondrocytes [100].Among the four receptors, the function of FGFR3 is the most well understood. Ablation of Fgfr3 in mice causes the expansion of proliferating and hypertrophic chondrocytes, whereas in humans, loss-of-function pathogenic variants in the FGFR3 gene could cause the camptodactyly, tall stature, scoliosis, and hearing loss (CATSHL) syndrome [101,102,103]. By contrast, the overexpression of Fgfr3 in mice and gain-of-function pathogenic variants in humans decrease the chondrocyte proliferation rate, leading to the development of achondroplasia [104].Fgfr3 is known to inhibit chondrocyte proliferation through increased expression of Snail1, that activates the signal transducer and activator of the transcription 1 (STAT1) pathway [105,106]. FGFR3-induced chondrocyte growth arrest is also dependent on the upregulation of cell-cycle inhibitors, such as p21 and p27, and requires the activated (phosphorylated) form of p107 and p130, but not pRb [107,108]. On the other side, conflicting data have been reported regarding its effect on chondrocyte maturation. Murakami et al. demonstrated that FGFR3 delays chondrocyte maturation through the MAP kinase (MAPK) pathway. In contrast, Minina and colleagues proved that, in mouse embryonic limb explants, FGF signaling acts upstream of the IHH/PTHrP and accelerates late steps of chondrocyte hypertrophy [109]. Furthermore, subsequent work by Dailey et al. demonstrated that the FGF treatment of rat chondrosarcoma chondrocytes modulates the gene expression profile to favor chondrogenic hypertrophy [110]. Altogether, these data suggest that FGF inhibits proliferation and the initial stage of hypertrophy, but then promotes maturation into late hypertrophic chondrocytes.
4. Bone Alterations in Mucopolysaccharidoses: From Patients to Animal Models
- Skeletal Alterations. MPSI and MPSII mice, as well as MPSI cats and dogs, present musculoskeletal deformities similar to those detected in affected patients [114,115,116,117]. In particular, they showed facial dysmorphisms, coxofemoral subluxation, fusion of the cervical vertebrae, pectus excavatum, and thickening of vertebrae arches. Shortened bones (femurs, tibiae, and lumbar vertebrae), a cardinal feature of human disease, were not observed in these models, in which bone lengths appeared similar to those of wild types [116,118]. In contrast, a reduction in bone length was detected in MPSVI rats and cats and in MPSVII dogs [119,120]. The MPSI and MPSVII canine models displayed lower bone mineral density compared to controls, similarly to what is observed in MPSI patients [121,122]. Conversely, mice models of MPSI and MPSVII showed higher bone mineral density, compared to their age-matched wild-type counterparts [118]. In particular, MPSI mice developed a progressive high bone mass phenotype, with a significantly reduced number of both osteoclasts and osteoblasts. Kuehn et al. investigated the activity of osteoclasts and their role in this high bone mass phenotype. Specifically, the authors demonstrated that BM transplantation from MPSI donor mice into wild-type recipient mice did not reproduce a high bone mass phenotype. Thus, they concluded that the increase in the trabecular bone could be due to impaired bone remodeling, determined by GAG accumulation in the bone matrix, rather than to defective osteoclast function [123].
- Growth plate morphological abnormalities. Both inbred strains and transgenic MPS animal models were able to also reproduce the abnormal growth plate morphology observed in the human disease, specifically, the disorganization of the columnar architecture in the proliferating and hypertrophic zones. Histological analysis of femurs of MPSI mice revealed thickening of the growth plate, in which the proliferative zone was thin and presented a disorganized distribution of chondrocytes [124,125]. In contrast, the hypertrophic zone was wider compared to healthy mice and showed an increased amount of large and swollen hypertrophic chondrocytes. Furthermore, it revealed that cartilage was retained within the mineralized cortical bone, and that the latter had lost its lamellar structure and organization [118,124].Abnormalities in chondrocytes, with enlarged lysosomes and growth plate disorganization, were also observed in MPSI dog and cat models, MPSVI cat models, and MPSIIIA, MPSIVA, and MPSVII mouse models [126,127,128]. In MPSVII dogs, lysosomes filled with storage materials were also detected in osteoblasts, osteoclasts, and osteocytes [129].
- Molecular consequences of GAG accumulation. GAGs are involved in multiple cellular pathways, through the regulation of the GF function and matrix release of cytokines and chemokines [130]. In addition, GAGs bind GF receptors, promoting conformational changes of the receptors themselves or of their ligands. Proteoglycans of the HS family can bind to a variety of growth factors, including members of the FGF family, bone morphogenic proteins (BMPs), transforming growth factor β (TGFβ), and WNT signaling pathways (reviewed in Bernfield et al.) [131]. It has been shown that HS-glycosaminoglycan is necessary in order for FGF ligands to bind to their cognate receptors [132]. However, the GAG excess in cells from MPSI patients results in the impairment of FGF/FGFR binding, and the FGF pathway activity was affected at an early stage of development and before the onset of bone defect in zebrafish and mice MPSII models [133,134]. GAG accumulation also impaired BMP-4 signaling in human multipotent adult progenitor cells derived from MPSI patients [135].Accumulation of HS in MPS patients does not occur in lysosomes only, but also affects other cellular and extracellular compartments. In MPSI and in MPSIIIB fibroblasts, extracellular accumulation of HS resulted in excessive binding of FGF2 and impaired activity of the growth factor, which was restored by reducing the excess HS [37].GAG affinity to growth factors is determined by their sulfation pattern [125]. Not only is the undegraded GAG accumulation involved in the pathogenesis of severe MPSI, but the abnormal GAG structure and sulfation pattern are involved as well. It has been shown in liver and brain tissues from 12-week-old MPSI mice that HS chains contain significantly increased N-, 2-O-, and 6-O-sulfation [136]. By contrast, cultured human BM-derived multipotent adult progenitor cells isolated from MPSI patients showed a reduction in 6-O sulfation of HS. This process results in impaired FGF-2 activity, as demonstrated by the fact that the replacement of normal HS on MPSI cells’ surfaces restored its function. The reduction in 6-O sulfation of HS in MPSI patients contributes to impaired FGF-2 activity, as demonstrated by the fact that the replacement of normal HS on MPSI cells’ surfaces restores its function [133]. In addition, it has been shown that GAG accumulation in MPSs could lead to growth plate anomalies, altering different processes involved in endochondral ossification, in particular, chondrocyte proliferation, as well as POC and SOC formation.How GAG accumulation in chondrocytes affects their proliferation capacity remains largely unknown. It has been shown that the growth plate of MPSVII mice exhibited a decreased chondrocyte proliferation, caused by increased activity of the anti-proliferative STAT1 and decreased activity of the pro-proliferative STAT3 factors [129,137]. In particular, STAT3 showed a markedly reduced phosphorylation at tyrosine705 as compared to normal mice, probably as the result of the diminished levels of leukemia inhibitory factor and IHH [137]. Recently, it has been shown that MPSVII chondrocytes are able to enter the cell cycle, but their ability to complete the cell cycle is impaired, thus leading to a delay in cellular proliferation and differentiation [129]. In MPSVII murine and canine models, abnormalities of chondrocytes were shown to be associated with a delayed formation of POC and SOC [138,139]. To explain this finding, it has been postulated that GAG accumulation in chondrocytes could alter the cellular processes involved in the transition of cartilage to bone, namely, hypertrophy, cartilage resorption, angiogenesis, and osteogenic differentiation. Peck et al. demonstrated the persistent expression of SOX9 in MPSVII dogs [139]. Since SOX9 directly represses the production of COL10A1, VEGF, and RUNX2 (see Section 3.1), its persistent expression could be responsible for the altered chondrocyte hypertrophy and the delayed bone formation. Indeed, the molecular profiling of epiphyseal tissues, isolated from 14-day-old MPSVII dogs, presented the downregulation of COL10A1, as well as RUNX2 and other genes important for osteogenic differentiation, such as alkaline phosphatase (ALPL), osteocalcin (BGLAP), and osteopontin (SPP1). In addition, MMP, WNT/beta catenin, and BMP pathways exhibited significantly altered overall expression [140]. MMPs are enzymes involved in cartilage degradation and are responsible for bone remodeling and angiogenesis. MMP13 is expressed in hypertrophic chondrocytes and, together with MMP9, facilitates angiogenesis and, subsequently, the migration of bone-forming cells, allowing the formation of POC and SOC [140]. Downregulation of Mmp13 has been observed also in MPSI mice [137]. Interestingly, MMP13 transcription is controlled by RUNX2, and therefore, it may be possible that the MMP13 downregulation in MPS chondrocytes could be an indirect consequence of persistent SOX9 expression [141].Finally, it has been demonstrated that MPSI mouse bones have an altered cathepsin K activity. Cathepsin K is a cysteine protease member of the cathepsin lysosomal protease family. It is highly expressed in osteoclasts, and it is responsible for collagen type II degradation. In MPSI mice, HS and DS accumulation blocks the catabolic activity of cathepsin K, thus resulting in a decreased cartilage resorption that contributes to the growth plate pathology of MPSI [142].
5. Therapeutic Options for MPSI and Focus on Bone Limitations
5.1. Hematopoietic Stem Cell Transplantation
5.2. Enzyme Replacement Therapy
5.3. Gene Therapy
- In vivo GT. The vector carrying the therapeutic gene can be directly injected in the case of in vivo GT. Being systemic or targeted to specific tissues, the administration of adeno-associated viruses (AAVs) could induce a long-term target gene expression, with a low genotoxicity risk [206]. The efficacy of this approach for treating MPSI has been demonstrated through many studies performed in animal models, with a better outcome observed when treatment was started early in life [207,208]. In particular, improvements in bone disease, craniofacial parameters, and neurological symptoms were obtained when AAVs were injected into neonatal mice [209]. For treating other MPSs, with the aim to reach bone and cartilage, AAVs have been modified in their capsid region for specifically targeting hydroxyapatite, a binding protein present in bone [210]. In treated mice, prolonged gene expression was observed in this targeted compartment with increased enzyme activity, suggesting that this approach could be translated to other MPSs with skeletal involvement.Starting from promising in vitro studies on MPSI fibroblasts, gene therapy using retroviruses (RVs) has been tested in several animal models [211,212,213]. First attempts showed the limited expression stability achieved and the loss over time, highlighting the need for pharmacological immunosuppression to limit the response of cytotoxic T lymphocytes and for gene expression avoidance in antigen-presenting cells [206,214]. When treating adult mice or young dogs with an alpha-1-antitrypsin promoter, many multisystemic manifestations and the cerebral storage were corrected without showing limiting auto-antibody formation [215]. In terms of skeletal defects, GT mice achieved mineralization normalization and a reduction in femur width, whereas no facial dysmorphism was observed in dogs after the early initiation of therapy [116,215].Ten years after RV-mediated in vivo GT, lentivirus (LV) efficacy was evaluated first on MPSI fibroblasts, where a long-term gene expression was achieved, and consequently, in animal models, in which even a little improvement in gene correction caused GAG reduction, despite the formation of anti-IDUA IgG limiting the long-term effect [37,216].
- Ex vivo GT. In the case of ex vivo GT application, autologous HSCs are employed for carrying the IDUA protein and inducing metabolic correction through the modified progenitors’ engraftment, differentiation, and release of the missing enzyme into local compartments [217]. Overall, after insertion into the genome, supraphysiological IDUA expression levels could be achieved, overcoming the ERT limitations of chronic injections. Moreover, an autologous approach, obtained by modifying patient-derived cells, is fundamental for reducing the immunological complications associated with standard HSCT.First attempts of ex vivo GT approaches were tested using RVs for transducing donor BM-derived cells and delivering the therapeutic gene in MPSI mice [218]. Visceral enzyme recovery and metabolic reduction were obtained months after treatment, with morphological improvements observed also in brain, but there was a lack of skeletal improvements. When erythroid cells were reprogrammed with targeting LVs, MPSI mice showed long-term enzymatic correction, vacuole reduction in organs, and cognitive improvement [219]; bone defect ameliorations were not investigated. Afterwards, Visigalli et al. demonstrated that results obtained through gene-corrected cells transplanted into adult MPSI mice highly exceeded the HSCT outcome thanks to the supranormal IDUA levels achieved in several tissues [220]. Metabolic and phenotypic defects in hard-to-treat organs were normalized with the amelioration of neurologic manifestations. When considering the skeletal compartment, femur density and dimension analyses, growth plate architecture evaluation, and computed tomography scans of long bones and of the zygomatic arch, values almost comparable to healthy ones were suggested after GT. No significant genotoxicity nor tumorigenic risks were associated with this treatment, as provided by safety preclinical studies that paved the way for the ongoing clinical trial at San Raffaele Hospital (NCT03488394) [221,222]. In this phase 1/2 study, autologous CD34+ cells were genetically modified with the LV encoding for the IDUA enzyme and transplanted in eight severe MPSI patients. Overall, this procedure was safe; all patients showed sustained engraftment of gene-corrected cells with blood IDUA activity reaching supraphysiologic levels after GT. Urinary GAG excretion levels reduced to normal or near-normal values by 1-year post-treatment. IDUA activity in the cerebrospinal fluid became detectable by month 3 post-GT in all subjects, accompanied by a progressive decrease in GAG storage. With a median follow-up of 2 years, patients showed stable cognitive and motor performances, reduced joint stiffness, improved or stable findings on brain and spine MRI, and normal growth according to peers (NCT03488394) [222]. These results highlighted the therapeutic potential of ex vivo GT for the treatment of severe MPSI.
- Gene editing. A specific gene could be potentially permanently modified for therapeutic purposes through gene editing approaches. The gene of interest could undergo editing, disruption, replacement, or addition in a particular site after double-strand breaks and DNA repair processes. In this context, nucleases are employed for editing the sequence, through the most commonly used zinc-finger nucleases (ZFNs), transcription activator-like endonucleases (TALENs), and CRISPR/Cas9 tools with targeting accuracy [223]. The complementary lacking gene portion, supplied with specific homology arms, could be provided and inserted by the homology direct-repair mechanism in a permanent manner [224]. In the case of a lack of the delivered sequence, non-homologous end-joining could induce a frame shift, with the addition or removal of bases in the proximity of the cut.In the case of MPS treatment, the target sequence and CRISPR/Cas9 machinery components were delivered through a liposome formulation into newborn MPSI mice; it allowed metabolic correction in some compartments, including in the cardiac tissue, with no CNS effect nor evaluation of bone amelioration [225]. Genome-edited human HSCs, modified through the CRISPR/Cas9 technology, were transplanted in adult immunodeficient MPSI mice and caused a limited increase in circulating IDUA, that was, however, enough for causing metabolic normalization of the main visceral organs and partial improvements in the CNS manifestations. This approach corrected the phenotypic bone alterations of treated mice, normalizing skull and long-bone thickness [226].On the other hand, novel genome editing strategies use delivered ZFN for inserting the therapeutic gene and inducing normal transgene expression. By targeting the albumin locus, positive preliminary preclinical results in visceral and neurological manifestations paved the way for its application in MPSI-attenuated patients. Since 2016, Sangamo Therapeutics started an ongoing phase 1/2 clinical study that shows a lack of severe side effects, but low gene expression (NCT02702115; NCT04628871) [227]. No indications about its effect on the skeletal outcome were reported up to now.
- Non-viral delivery methods. Together with AAV-, RV-, and LV-based delivery of the therapeutic gene, non-viral methods have been applied for the treatment of MPSI. By using a hydrodynamic technique for targeting the liver and transposing the cassette of interest into the genome, the Sleeping Beauty (SB) transposon system represents one of the most applied non-viral approaches for delivering the therapeutic transgene. Its application for MPSI treatment has demonstrated an effect in reducing biochemical and metabolic defects in mice [228]. In terms of skeletal defects, SB treatment caused a reduction in zygomatic thickness, with no growth plate defects observed in long bones and a loss of swollen cells in the BM [228]. No improvements in cortical thickness or mid-diaphyseal bone regions’ dimensions were reported.
5.4. Potential Alternative Approaches for MPSI Treatment (Table 1)
- Storage reduction strategies. More precise studies of the pathophysiology behind MPS disorders have encouraged the investigation of several small-molecule therapies. In particular, storage reduction approaches act on blocking GAG production, instead of promoting their catabolism, by balancing synthesis over degradation. Overall, in contrast to ERT, the idea is to avoid GAG synthesis and disease worsening, and a therapeutic benefit for skeletal manifestations could also be possible [231,232].Genistein (5,7-dihydroxy-3-[4-hydroxyphenyl]-4H-1-benzopyran-4-one) is an isoflavone that binds and inactivates the epidermal growth factor receptor, which is fundamental for GAG formation [233]. When tested in MPSII and III patients (PO 5 mg/kg/day), improvements in some clinical and behavioral aspects were observed, likely due to Genistein’s ability to cross the blood–brain barrier [234,235]. Despite articular mobility that resulted in being ameliorated when administered, further clinical efficacy is still to be determined, especially considering side effects observed in treated mice that are probably due to a broader effect of the molecule on different pathways [234,235,236].Alternative strategies include the administration of small compounds for reducing secondary molecules, such as gangliosides, whose synthesis could be blocked using Miglustat [237]. On the other hand, short interfering RNA (shRNA) molecules could be applied for silencing genes that encode for enzymes that take part in GAG metabolism [238]. This approach was tested in MPSI fibroblasts and caused reduced synthesis, highlighting its possible application for MPS treatment.
- Nonsense suppression treatment. In MPSI patients with a genotype characterized by the presence of nonsense pathogenic variants in the IDUA gene, nonsense suppression therapies could be used. Specific molecules could promote the bypassing of the termination codon, allowing elongation to proceed, and subsequently, allow for the production of a full-length protein that retains its enzymatic properties. Additionally, a partial enzyme restoration could significantly ameliorate disease manifestations, considering MPSI as a promising candidate. The combination of nonsense-mediated mRNA decay attenuation and suppression treatments, such as gentamicin or NB84, was tested in MPSI mice, resulting in beneficial effects without toxicity, restoring the enzymatic activity, and reducing GAG accumulation [239]. The progression of skeletal involvement, together with cardiac and cerebral hard-to-treat compartments, was delayed after long-term treatment with NB84 in MPSI animals, as demonstrated by both improved architecture parameters and osteoclast number reduction [240]. These promising results promoted these alternative applications for treating MPS patients, although toxicity remains a limiting factor.
- Chaperone therapy. Chaperones could represent a valid treatment option in case of mutations that impair protein folding and consequent trafficking and accumulation. This therapy stabilizes these misfolded proteins by mainly rearranging their hydrophobic portion, and avoids storage and degradation, since enzyme activity and trafficking have been corrected [241]. Differently from ERT, the CNS and several organs could be reached, along with no immunogenicity and administration ease. Nonetheless, despite the application for many MPSs, a restriction to specific mutations, off-target toxicity, and enzyme inhibition risk could limit its applicability [241,242].
- Others. New strategies for treating MPSs are currently under investigation. Some preliminary in vitro results have been obtained after overexpression of the TFEB (transcription factor EB) gene, which is important for the autophagy regulation that is severely affected in MPSs. Altering the autophagic-lysosomal pathway in MPSIIIA resulted in the clearance of GAG accumulation [243].
5.5. Alternative Treatments Focused on Skeletal Defects
- Anti-inflammatory drugs. Since inflammation highly regulates MPSI pathogenesis and inflammatory pathways are activated in affected patients, targeting inflammation with anti-inflammatory drugs could limit its effects on connective tissue alterations and disease progression.Pentosan polysulfate (PPS), a molecule with prochondral activity and anti-inflammatory properties, was tested in the canine model of MPSI and noted to cause an overall decrease in GAG accumulation [252]. Similarly, adult MPS patients that received the PPS therapy combined with the standard ERT showed improvements in hip, knee, and ankle mobility, suggesting that inflammatory impairment plays a crucial role in MPSI [253]. Alternatively, infliximab, with its tumor necrosis factor (TNF)α-binding properties and effect on the TLR4–TNFα axis, was injected in MPS animals, and therapeutic benefits were appreciated when combined with standard ERT [254].Additionally, the TNF inhibitor adalimumab is currently under investigation in a phase 1/2 study; its effect on the dysostosis multiplex manifestations is being tested in MPSI patients, together with safety and efficacy analyses (NCT03153319).Combinational approaches to standard therapies could substitute ERT and avoid antibody formation in the case of early treatment after newborn screening, with the final goal of treating younger patients [252].
- The use of MMPs and apoptosis inhibitors, cytokines, and WNT/β-catenin agonists could represent an alternative approach for counteracting the bone defects observed in many MPSs [48]. In particular, Simonaro et al. demonstrated that matrix metalloproteinases MPP2 and MMP9 were impaired, causing matrix and bone formation alterations, and that reduced mineralization resulted from defects in hypertrophic chondrocytes. In addition, the high level of cartilage death was compensated for by TGFβ release to increase the proliferation rate. Cytokines and MPPs highly moderate the pathways involved in skeletal balance, and targeting these key players could be a valuable therapeutic approach to be combined with standard treatments, with the aim of achieving the correct bone formation and maturation, especially in young patients.
- Growth factor therapy. GAG accumulation severely impairs the bone milieu and the released growth factors’ role, contributing to MPSI skeletal degeneration. By knowing the specific interaction and function of the GF in the context of bone and other organs, targeting specific receptors could be an alternative strategy to couple with standard approaches [125]. A combined approach using C-type natriuretic peptide and ERT, tested in MPSVII mice for promoting growth improvement, resulted in a synergic effect that overcame the two monotherapies; regulating chondrocyte apoptosis on one side and thickening of the growth plate on the other caused amelioration of the hypertrophic zone composition and, overall, articular cartilage growth, suggesting that the skeletal defects of MPSs could benefit from this combined strategy [255].Recent studies have highlighted the fine regulation of the mammalian target of rapamycin complex 1 (mTORC1) signaling for appropriate longitudinal skeletal growth; in particular, impaired collagen trafficking in osteoclasts was hypothesized in the case of MPS mice, with delayed secretion due to unbalanced collagen metabolism. Altered TORC1-dependent post-translational modifications in UV radiation resistance-associated (UVRAG) protein were observed in patient-derived chondrocytes and precursors, which were isolated from MPSVI and MPSI primary samples, respectively. Bone formation was restored after a reduction in mTORC1 signaling or Tat-Beclin1, a peptide able to induce autophagy, overall improving the collagen composition in cartilages in MPSVI and MPSVII mice [256].
- Abnormal osteoactivin expression was found in some MPSs. Osteoactivin has the fundamental role of regulating endochondral ossification, proper osteoblast differentiation, and autophagy. Acting on this pathway could represent a specific therapeutic approach for ameliorating the bone involvement in MPSI patients [140].
- MSCs represent potential therapeutic targets because of their role in immunomodulation, regeneration, and osteoblast or chondrocyte differentiation, especially when considering combined approaches for MPSI. Positive effects on motor coordination were observed when healthy BM-derived multipotent stem cells were intracerebroventricularly injected in affected mice [257]. Modified MSCs were successfully able to normalize enzyme activity and retinal defects in MPS mice when precociously treated, highlighting the feasibility of neonatal ex vivo GT [258]. When injected in patients with severe MPSI, bone mineral density was maintained or slightly improved, probably due to the already set skeletal defects and late time of intervention [259]. The MSC effect on precociously halting dysostosis multiplex progression needs to be evaluated.
- Biphosphonates have been considered as a supportive therapy for MPSs in light of their ability to counteract bone resorption by impairing several osteoclast activities, promoting osteoclast apoptosis, and increasing bone mass [260]. In particular, neridronate given intravenously has been reported to improve functional bone performance and the radiological phenotype of a patient affected by MPSIVA [261].
5.6. Early Treatment of MPSI Disease Manifestations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sakuru, R.; Bollu, P.C. Hurler Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; De Jong, J.G.N.; Van Weely, S.; Niezen-Koning, K.E.; Van Diggelen, O.P. The frequency of lysosomal storage diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef]
- Gifford, S.R.; Scheie, H.G.; Hambrick, G.W.; Barness, L.A. A Newly Recognized Forme Fruste of Hurler’s Disease (Gargoylism)* *From the Departments of Ophthalmology, Dermatology, and Pediatrics, Hospital of the University of Pennsylvania, Children’s Hospital of Philadelphia, and University of Pennsylvania Medical School. Am. J. Ophthalmol. 1962, 53, 753–769. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. The international consensus panel on the management and treatment of mucopolysaccharidosis I mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Blattner, R.J. Hurler’s syndrome. J. Pediatr. 1966, 69, 313–315. [Google Scholar] [CrossRef]
- Muenzer, J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J. Pediatr. 2004, 144, S27–S34. [Google Scholar] [CrossRef]
- Available online: https://Databases.Lovd.Nl/Shared/Genes/IDUA (accessed on 16 September 2022).
- Jahic, A.; Günther, S.; Muschol, N.; Fossøy Stadheim, B.; Braaten, Ø.; Kjensli Hyldebrandt, H.; Kuiper, G.; Tylee, K.; Wijburg, F.A.; Beetz, C. “Missing mutations” in MPS I: Identification of two novel copy number variations by an IDUA -specific in house MLPA assay. Mol. Genet. Genom. Med. 2019, 7, e00615. [Google Scholar] [CrossRef] [PubMed]
- Çelik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef]
- Voskoboeva, E.Y.; Bookina, T.M.; Semyachkina, A.N.; Mikhaylova, S.V.; Vashakmadze, N.D.; Baydakova, G.V.; Zakharova, E.Y.; Kutsev, S.I. Mucopolysaccharidosis Type I in the Russian Federation and Other Republics of the Former Soviet Union: Molecular Genetic Analysis and Epidemiology. Front. Mol. Biosci. 2022, 8, 783644. [Google Scholar] [CrossRef] [PubMed]
- Bunge, S.; Kleijer, W.J.; Steglich, C.; Zuther, C.; Morris, C.; Beck, M.; Schwinger, E.; Hopwood, J.J.; Scott, H.S.; Gal, A. Mucopolysaccharidosis type I: Identification of 8 novel mutations and determination of the frequency of the two common α-L-iduronidase mutations (W402X and Q70X) among European patients. Hum. Mol. Genet. 1994, 3, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Voskoboeva, E.Y.; Krasnopolskaya, X.D.; Mirenburg, T.V.; Weber, B.; Hopwood, J.J. Molecular Genetics of Mucopolysaccharidosis Type I: Mutation Analysis among the Patients of the Former Soviet Union. Mol. Genet. Metab. 1998, 65, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef]
- Alif, N.; Hess, K.; Straczek, J.; Sebbar, S.; N’Bou, A.; Nabet, P.; Dousset, B. Mucopolysaccharidosis type I: Characterization of a common mutation that causes Hurler syndrome in Moroccan subjects. Ann. Hum. Genet. 1999, 63, 9–16. [Google Scholar] [CrossRef]
- Chkioua, L.; Khedhiri, S.; Kassab, A.; Bibi, A.; Ferchichi, S.; Froissart, R.; Vianey-Saban, C.; Laradi, S.; Miled, A. Molecular analysis of mucopolysaccharidosis type I in Tunisia: Identification of novel mutation and eight Novel polymorphisms. Diagn. Pathol. 2011, 6, 39. [Google Scholar] [CrossRef]
- Tebani, A.; Zanoutene-Cheriet, L.; Adjtoutah, Z.; Abily-Donval, L.; Brasse-Lagnel, C.; Laquerrière, A.; Marret, S.; Benabdellah, A.C.; Bekri, S. Clinical and Molecular Characterization of Patients with Mucopolysaccharidosis Type I in an Algerian Series. Int. J. Mol. Sci. 2016, 17, 743. [Google Scholar] [CrossRef]
- Ficicioglu, C.; Giugliani, R.; Harmatz, P.; Mendelsohn, N.J.; Jego, V.; Parini, R. Intrafamilial variability in the clinical manifestations of mucopolysaccharidosis type II: Data from the Hunter Outcome Survey (HOS). Am. J. Med. Genet. Part A 2018, 176, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Frigeni, M.; Rodriguez-Buritica, D.F.; Saavedra, H.; Gunther, K.A.; Hillman, P.R.; Balaguru, D.; Northrup, H. The youngest pair of siblings with Mucopolysaccharidosis type IVA to receive enzyme replacement therapy to date: A case report. Am. J. Med. Genet. Part A 2021, 185, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef]
- Giugliani, R.; Muschol, N.; Keenan, H.A.; Dant, M.; Muenzer, J. Improvement in time to treatment, but not time to diagnosis, in patients with mucopolysaccharidosis type I. Arch. Dis. Child. 2021, 106, 674–679. [Google Scholar] [CrossRef]
- Sheth, J.; Mistri, M.; Shah, K.; Chaudhary, M.; Godbole, K.; Sheth, F. Lysosomal Storage Disorders in Nonimmune Hydrops Fetalis (NIHF): An Indian Experience. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 35, pp. 47–52. ISBN 978-3-662-55832-4. [Google Scholar]
- Kuiper, G.-A.; Meijer, O.L.M.; Langereis, E.J.; Wijburg, F.A. Failure to shorten the diagnostic delay in two ultra-orphan diseases (mucopolysaccharidosis types I and III): Potential causes and implications. Orphanet J. Rare Dis. 2018, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef]
- de Ru, M.H.; Teunissen, Q.G.; van der Lee, J.H.; Beck, M.; Bodamer, O.A.; Clarke, L.A.; Hollak, C.E.; Lin, S.-P.; Rojas, M.-V.M.; Pastores, G.M.; et al. Capturing phenotypic heterogeneity in MPS I: Results of an international consensus procedure. Orphanet J. Rare Dis. 2012, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef]
- Cleary, M.A.; Wraith, J.E. The presenting features of mucopolysaccharidosis type IH (Hurler syndrome). Acta Paediatr. 1995, 84, 337–339. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Whitley, C.B.; Eisengart, J.B. Beneath the floor: Re-analysis of neurodevelopmental outcomes in untreated Hurler syndrome. Orphanet J. Rare Dis. 2018, 13, 76. [Google Scholar] [CrossRef]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early diagnostic signs in infants and children. Ital. J. Pediatr. 2018, 44, 133. [Google Scholar] [CrossRef]
- Moore, D.; Connock, M.J.; Wraith, E.; Lavery, C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J. Rare Dis. 2008, 3, 24. [Google Scholar] [CrossRef]
- Schmidt, H.; Ullrich, K.; Von Lengerke, H.-J.; Kleine, M.; Brämswig, J. Radiological findings in patients with mucopolysaccharidosis I H/S (Hurler-Scheie syndrome). Pediatr. Radiol. 1987, 17, 409–414. [Google Scholar] [CrossRef]
- Viskochil, D.; Clarke, L.A.; Bay, L.; Keenan, H.; Muenzer, J.; Guffon, N. Growth patterns for untreated individuals with MPS I: Report from the international MPS I registry. Am. J. Med. Genet. A 2019, 179, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Bentley, A.; Chen, C.; Dhemech, M.W.; Gee, E.; Orton, P.; Pringle, C.; Rajan, J.; Saxena, A.; Tol, G.; et al. Non-cardiac Manifestations in Adult Patients with Mucopolysaccharidosis. Front. Cardiovasc. Med. 2022, 9, 839391. [Google Scholar] [CrossRef]
- Amendum, P.C.; Khan, S.; Yamaguchi, S.; Kobayashi, H.; Ago, Y.; Suzuki, Y.; Celik, B.; Rintz, E.; Hossain, J.; Xiao, W.; et al. Glycosaminoglycans as Biomarkers for Mucopolysaccharidoses and Other Disorders. Diagnostics 2021, 11, 1563. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, V.; Sarogni, P.; Pistorio, V.; Cerulo, G.; Paladino, S.; Pavone, L.M. Targeting Heparan Sulfate Proteoglycans as a Novel Therapeutic Strategy for Mucopolysaccharidoses. Mol. Ther.-Methods Clin. Dev. 2018, 10, 8–16. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; De Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Dermatan sulfate and heparan sulfate as a biomarker for mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Tomatsu, S. Advances in glycosaminoglycan detection. Mol. Genet. Metab. 2020, 130, 101–109. [Google Scholar] [CrossRef]
- Olczyk, K. Age-related changes in glycosaminoglycans of human intervertebral discs. Folia Histochem. Cytobiol. 1993, 31, 215–220. [Google Scholar]
- Lawrence, R.; Prill, H.; Vachali, P.P.; Adintori, E.G.; De Hart, G.; Wang, R.Y.; Burton, B.K.; Pasquali, M.; Crawford, B.E. Characterization of disease-specific chondroitin sulfate nonreducing end accumulation in mucopolysaccharidosis IVA. Glycobiology 2020, 30, 433–445. [Google Scholar] [CrossRef]
- Saunders, S.; Bernfield, M. Cell surface proteoglycan binds mouse mammary epithelial cells to fibronectin and behaves as a receptor for interstitial matrix. J. Cell Biol. 1988, 106, 423–430. [Google Scholar] [CrossRef]
- Murdoch, A.D.; Dodge, G.R.; Cohen, I.; Tuan, R.S.; Iozzo, R.V. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 1992, 267, 8544–8557. [Google Scholar] [CrossRef] [PubMed]
- Johanna, W.C.; Celie, J.W.A.M.; Beelen, R.H.J.; Born, J.V.D. Heparan sulfate proteoglycans in extravasation: Assisting leukocyte guidance. Front. Biosci. 2009, 14, 4932–4949. [Google Scholar] [CrossRef]
- Settembre, C.; Cinque, L.; Bartolomeo, R.; Di Malta, C.; De Leonibus, C.; Forrester, A. Defective collagen proteostasis and matrix formation in the pathogenesis of lysosomal storage disorders. Matrix Biol. 2018, 71–72, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and Bone Disease in Mucopolysaccharidoses VI and VII: Identification of New Therapeutic Targets and BioMarkers Using Animal Models. Pediatr. Res. 2005, 57, 701–707. [Google Scholar] [CrossRef]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of Glycosaminoglycan-Mediated Bone and Joint Disease: Implications for the Mucopolysaccharidoses and Other Connective Tissue Diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.-M.; Sommer, B.; et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front. Cell Dev. Biol. 2017, 5, 106. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An Endosomal NAADP-Sensitive Two-Pore Ca 2+ Channel Regulates ER-Endosome Membrane Contact Sites to Control Growth Factor Signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.L.; De Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Karsenty, G.; Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 2013, 12, 2744–2752. [Google Scholar] [CrossRef]
- Nabavi, N.; Urukova, Y.; Cardelli, M.; Aubin, J.E.; Harrison, R.E. Lysosome Dispersion in Osteoblasts Accommodates Enhanced Collagen Production during Differentiation. J. Biol. Chem. 2008, 283, 19678–19690. [Google Scholar] [CrossRef] [PubMed]
- Tsukuba, T.; Sakai, E.; Nishishita, K.; Kadowaki, T.; Okamoto, K. New functions of lysosomes in bone cells. J. Oral Biosci. 2017, 59, 92–95. [Google Scholar] [CrossRef]
- Fernández, B.; Fdez, E.; Gómez-Suaga, P.; Gil, F.; Molina-Villalba, I.; Ferrer, I.; Patel, S.; Churchill, G.C.; Hilfiker, S. Iron overload causes endolysosomal deficits modulated by NAADP-regulated 2-pore channels and RAB7A. Autophagy 2016, 12, 1487–1506. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.G.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Reolon, G.K.; Reinke, A.; de Oliveira, M.R.; Braga, L.M.; Camassola, M.; Andrades, M.; Moreira, J.C.F.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in Oxidative Markers in the Cerebellum and Peripheral Organs in MPS I Mice. Cell. Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in Mouse Models of Mucopolysaccharidosis Type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [PubMed]
- Thorogood, P.V.; Hinchliffe, J.R. An analysis of the condensation process during chondrogenesis in the embryonic chick hind limb. Development 1975, 33, 581–606. [Google Scholar] [CrossRef]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S.E. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the Endochondral Skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef]
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M. Osteoblast Precursors, but Not Mature Osteoblasts, Move into Developing and Fractured Bones along with Invading Blood Vessels. Dev. Cell 2010, 19, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef]
- Lefebvre, V.; Li, P.; De Crombrugghe, B. A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998, 17, 5718–5733. [Google Scholar] [CrossRef]
- Hata, K.; Takahata, Y.; Murakami, T.; Nishimura, R. Transcriptional Network Controlling Endochondral Ossification. J. Bone Metab. 2017, 24, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zuscik, M.J.; Hilton, M.J.; Zhang, X.; Chen, D.; O’Keefe, R.J. Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Investig. 2008, 118, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Hargrave, M.R.; Christiansen, J.; Cooper, L.; Kun, J.; Evans, T.; Gangadharan, U.; Greenfield, A.; Koopman, P. The Sty-Related Gene Sox9 Is Expressed during Chondrogenesis in Mouse Embryos. Nat. Genet. 1995, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.M.; Deng, J.M.; Zhang, Z.P.; Behringer, R.R.; De Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Müller, C.; Gebhard, S.; Bauer, E.; Pausch, F.; Schlund, B.; Bösl, M.R.; Hess, A.; Surmann-Schmitt, C.; von der Mark, H.; et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 2010, 137, 901–911. [Google Scholar] [CrossRef]
- Leung, V.Y.L.; Gao, B.; Leung, K.K.H.; Melhado, I.G.; Wynn, S.L.; Au, T.Y.K.; Dung, N.W.F.; Lau, J.Y.B.; Mak, A.C.Y.; Chan, D.; et al. SOX9 Governs Differentiation Stage-Specific Gene Expression in Growth Plate Chondrocytes via Direct Concomitant Transactivation and Repression. PLoS Genet. 2011, 7, e1002356. [Google Scholar] [CrossRef] [PubMed]
- Dy, P.; Wang, W.; Bhattaram, P.; Wang, Q.; Wang, L.; Ballock, R.T.; Lefebvre, V. Sox9 Directs Hypertrophic Maturation and Blocks Osteoblast Differentiation of Growth Plate Chondrocytes. Dev. Cell 2012, 22, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Topol, L.; Lee, H.; Wu, J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development 2003, 130, 1003–1015. [Google Scholar] [CrossRef]
- Akiyama, H.; Lyons, J.P.; Mori-Akiyama, Y.; Yang, X.; Zhang, R.; Zhang, Z.; Deng, J.M.; Taketo, M.M.; Nakamura, T.; Behringer, R.R.; et al. Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Topol, L.; Chen, W.; Song, H.; Day, T.F.; Yang, Y. Sox9 Inhibits Wnt Signaling by Promoting β-Catenin Phosphorylation in the Nucleus. J. Biol. Chem. 2009, 284, 3323–3333. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Zhou, G.; Zheng, Q.; Engin, F.; Munivez, E.; Chen, Y.; Sebald, E.; Krakow, D.; Lee, B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19004–19009. [Google Scholar] [CrossRef]
- Cheng, A.; Genever, P.G. SOX9 determines RUNX2 transactivity by directing intracellular degradation. J. Bone Miner. Res. 2010, 25, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of Rate of Cartilage Differentiation by Indian Hedgehog and PTH-Related Protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.K.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP Receptor in Early Development and Indian Hedgehog—Regulated Bone Growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Chung, U.-I.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Investig. 2001, 107, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Soegiarto, D.W.; Yang, Y.; Lanske, B.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J. Clin. Investig. 2005, 115, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Hilton, M.J.; Tu, X.; Long, F. Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev. Biol. 2007, 308, 93–105. [Google Scholar] [CrossRef]
- Maeda, Y.; Schipani, E.; Densmore, M.J.; Lanske, B. Partial rescue of postnatal growth plate abnormalities in Ihh mutants by expression of a constitutively active PTH/PTHrP receptor. Bone 2010, 46, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Karp, S.J.; Schipani, E.; St-Jacques, B.; Hunzelman, J.; Kronenberg, H.; McMahon, A.P. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein-dependent and -independent pathways. Development 2000, 127, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.C.; Philbrick, W.M.; Amling, M.; Neff, L.A.; Baron, R.; Broadus, A.E. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc. Natl. Acad. Sci. USA 1996, 93, 10240–10245. [Google Scholar] [CrossRef]
- Lee, K.; Lanske, B.; Karaplis, A.C.; Deeds, J.D.; Kohno, H.; Nissenson, R.A.; Kronenberg, H.M.; Segre, G.V. Parathyroid hormone-related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology 1996, 137, 5109–5118. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Cohen, T.; Yao, T.-P.; Lassar, A.B. Parathyroid Hormone-Related Peptide Represses Chondrocyte Hypertrophy through a Protein Phosphatase 2A/Histone Deacetylase 4/MEF2 Pathway. Mol. Cell. Biol. 2009, 29, 5751–5762. [Google Scholar] [CrossRef]
- Correa, D.; Hesse, E.; Seriwatanachai, D.; Kiviranta, R.; Saito, H.; Yamana, K.; Neff, L.; Atfi, A.; Coillard, L.; Sitara, D.; et al. Zfp521 Is a Target Gene and Key Effector of Parathyroid Hormone-Related Peptide Signaling in Growth Plate Chondrocytes. Dev. Cell 2010, 19, 533–546. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, X.; Lefebvre, V. Phosphorylation of SOX9 by Cyclic AMP-Dependent Protein Kinase A Enhances SOX9’s Ability To Transactivate ACol2a1 Chondrocyte-Specific Enhancer. Mol. Cell. Biol. 2000, 20, 4149–4158. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002, 16, 1446–1465. [Google Scholar] [CrossRef]
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast Growth Factor Receptor 3 Is a Negative Regulator of Bone Growth. Cell 1996, 84, 911–921. [Google Scholar] [CrossRef]
- Colvin, J.S.; Bohne, B.A.; Harding, G.W.; McEwen, D.G.; Ornitz, D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 1996, 12, 390–397. [Google Scholar] [CrossRef]
- Toydemir, R.M.; Brassington, A.E.; Bayrak-Toydemir, P.; Krakowiak, P.A.; Jorde, L.B.; Whitby, F.G.; Longo, N.; Viskochil, D.H.; Carey, J.C.; Bamshad, M.J. A Novel Mutation in FGFR3 Causes Camptodactyly, Tall Stature, and Hearing Loss (CATSHL) Syndrome. Am. J. Hum. Genet. 2006, 79, 935–941. [Google Scholar] [CrossRef]
- Naski, M.C.; Colvin, J.S.; Coffin, J.D.; Ornitz, D. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 1998, 125, 4977–4988. [Google Scholar] [CrossRef]
- Sahni, M.; Ambrosetti, D.-C.; Mansukhani, A.; Gertner, R.; Levy, D.; Basilico, C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999, 13, 1361–1366. [Google Scholar] [CrossRef]
- de Frutos, C.A.; Vega, S.; Manzanares, M.; Flores, J.M.; Huertas, H.; Martínez-Frías, M.L.; Nieto, M.A. Snail1 Is a Transcriptional Effector of FGFR3 Signaling during Chondrogenesis and Achondroplasias. Dev. Cell 2007, 13, 872–883. [Google Scholar] [CrossRef]
- Krejci, P.; Bryja, V.; Pachernik, J.; Hampl, A.; Pogue, R.; Mekikian, P.; Wilcox, W.R. FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp. Cell Res. 2004, 297, 152–164. [Google Scholar] [CrossRef]
- Laplantine, E.; Rossi, F.; Sahni, M.; Basilico, C.; Cobrinik, D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J. Cell Biol. 2002, 158, 741–750. [Google Scholar] [CrossRef]
- Minina, E.; Kreschel, C.; Naski, M.C.; Ornitz, D.M.; Vortkamp, A. Interaction of FGF, Ihh/Pthlh, and BMP Signaling Integrates Chondrocyte Proliferation and Hypertrophic Differentiation. Dev. Cell 2002, 3, 439–449. [Google Scholar] [CrossRef]
- Dailey, L.; Laplantine, E.; Priore, R.; Basilico, C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J. Cell Biol. 2003, 161, 1053–1066. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Shih, S.-C.; Chuang, C.-K.; Chen, M.-R.; Niu, D.-M.; Lin, S.-P. Assessment of bone mineral density by dual energy x-ray absorptiometry in patients with mucopolysaccharidoses. Orphanet J. Rare Dis. 2013, 8, 71. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Thomas, W.; Fung, E.; Viskochil, D.; Stevenson, D.A.; Steinberger, J.; Orchard, P.; Whitley, C.B.; Ensrud, K.E. Low Bone Mineral Content and Challenges in Interpretation of Dual-Energy X-Ray Absorptiometry in Children with Mucopolysaccharidosis Types I, II, and VI. J. Clin. Densitom. 2014, 17, 200–206. [Google Scholar] [CrossRef]
- Silveri, C.P.; Kaplan, F.S.; Fallon, M.D.; Bayever, E.; August, C.S. Hurler syndrome with special reference to histologic abnormalities of the growth plate. Clin. Orthop. Relat. Res. 1991, 269, 305–311. [Google Scholar] [CrossRef]
- Clarke, L.A.; Russell, C.S.; Pownall, S.; Warrington, C.L.; Borowski, A.; Dimmick, J.E.; Toone, J.; Jirik, F.R. Murine Mucopolysaccharidosis Type I: Targeted Disruption of the Murine α-L-Iduronidase Gene. Hum. Mol. Genet. 1997, 6, 9. [Google Scholar] [CrossRef]
- Haskins, M.E.; Jezyk, P.F.; Desnick, R.J.; Mcdonough, S.K.; Patterson, D.F. Alpha-L-iduronidase Deficiency in a Cat: A Model of Mucopolysaccharidosis I. Pediatr. Res. 1979, 13, 1294–1297. [Google Scholar] [CrossRef]
- Herati, R.S.; Knox, V.W.; O’Donnell, P.; D’Angelo, M.; Haskins, M.E.; Ponder, K.P. Radiographic evaluation of bones and joints in mucopolysaccharidosis I and VII dogs after neonatal gene therapy. Mol. Genet. Metab. 2008, 95, 142–151. [Google Scholar] [CrossRef][Green Version]
- Mansour, T.A.; Woolard, K.D.; Vernau, K.L.; Ancona, D.M.; Thomasy, S.M.; Sebbag, L.; Moore, B.A.; Knipe, M.F.; Seada, H.A.; Cowan, T.M.; et al. Whole genome sequencing for mutation discovery in a single case of lysosomal storage disease (MPS type 1) in the dog. Sci. Rep. 2020, 10, 6558. [Google Scholar] [CrossRef] [PubMed]
- Rowan, D.J.; Tomatsu, S.; Grubb, J.H.; Montaño, A.M.; Sly, W.S. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J. Inherit. Metab. Dis. 2013, 36, 235–246. [Google Scholar] [CrossRef]
- Yoshida, M.; Noguchi, J.; Ikadai, H.; Takahashi, M.; Nagase, S. Arylsulfatase B-deficient mucopolysaccharidosis in rats. J. Clin. Investig. 1993, 91, 1099–1104. [Google Scholar] [CrossRef]
- Dombrowski, D.C.S.; Carmichael, K.P.; Wang, P.; O’Malley, T.M.; Haskins, M.E.; Giger, U. Mucopolysaccharidosis type VII in a German Shepherd Dog. J. Am. Veter. Med. Assoc. 2004, 224, 553–557. [Google Scholar] [CrossRef]
- Chiaro, J.A.; Baron, M.D.; del Alcazar, C.M.; O’Donnell, P.; Shore, E.M.; Elliott, D.M.; Ponder, K.P.; Haskins, M.E.; Smith, L.J. Postnatal progression of bone disease in the cervical spines of mucopolysaccharidosis I dogs. Bone 2013, 55, 78–83. [Google Scholar] [CrossRef][Green Version]
- Peck, S.H.; Lau, Y.K.; Kang, J.L.; Lin, M.; Arginteanu, T.; Matalon, D.R.; Bendigo, J.R.; O’Donnell, P.; Haskins, M.E.; Casal, M.L.; et al. Progression of vertebral bone disease in mucopolysaccharidosis VII dogs from birth to skeletal maturity. Mol. Genet. Metab. 2021, 133, 378–385. [Google Scholar] [CrossRef]
- Kuehn, S.C.; Koehne, T.; Cornils, K.; Markmann, S.; Riedel, C.; Pestka, J.M.; Schweizer, M.; Baldauf, C.; Yorgan, T.A.; Krause, M.; et al. Impaired bone remodeling and its correction by combination therapy in a mouse model of mucopolysaccharidosis-I. Hum. Mol. Genet. 2015, 24, 7075–7086. [Google Scholar] [CrossRef]
- Russell, C.; Hendson, G.; Jevon, G.; Matlock, T.; Yu, J.; Aklujkar, M.; Ng, K.-Y.; Clarke, L.A. Murine MPS I: Insights into the pathogenesis of Hurler syndrome. Clin. Genet. 1998, 53, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Kingma, S.D.K.; Wagemans, T.; Ijlst, L.; Bronckers, A.L.J.J.; van Kuppevelt, T.H.; Everts, V.; Wijburg, F.A.; van Vlies, N. Altered interaction and distribution of glycosaminoglycans and growth factors in mucopolysaccharidosis type I bone disease. Bone 2016, 88, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Breider, M.A.; Shull, R.M.; Constantopoulos, G. Long-term effects of bone marrow transplantation in dogs with mucopolysaccharidosis I. Am. J. Pathol. 1989, 134, 677–692. [Google Scholar] [PubMed]
- Haskins, M.E.; McGrath, J.T. Meningiomas in Young Cats with Mucopolysaccharidosis I. J. Neuropathol. Exp. Neurol. 1983, 42, 664–670. [Google Scholar] [CrossRef]
- Frohbergh, M.; Ge, Y.; Meng, F.; Karabul, N.; Solyom, A.; Lai, A.; Iatridis, J.; Schuchman, E.H.; Simonaro, C.M. Dose Responsive Effects of Subcutaneous Pentosan Polysulfate Injection in Mucopolysaccharidosis Type VI Rats and Comparison to Oral Treatment. PLoS ONE 2014, 9, e100882. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Derrick-Roberts, A.L.K.; Reichstein, C.; Byers, S. Cell cycle progression is disrupted in murine MPS VII growth plate leading to reduced chondrocyte proliferation and transition to hypertrophy. Bone 2020, 132, 115195. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural Insights into Biological Roles of Protein-Glycosaminoglycan Interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Venkataraman, G.; Ernst, S.; Sasisekharan, V.; Sasisekharan, R. Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. FGF signaling deregulation is associated with early developmental skeletal defects in animal models for mucopolysaccharidosis type II (MPSII). Hum. Mol. Genet. 2018, 27, 2262–2275, Erratum in 2018, 27, 2407. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Nelson, M.S.; Pan, C.; Gaffney, P.M.; Gupta, P. Endogenous heparan sulfate and heparin modulate bone morphogenetic protein-4 signaling and activity. Am. J. Physiol. Cell Physiol. 2008, 294, C1387–C1397. [Google Scholar] [CrossRef] [PubMed]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñonuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis Type I, Unique Structure of Accumulated Heparan Sulfate and Increased N-Sulfotransferase Activity in Mice Lacking α-l-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.A.; Zhang, Y.; Hilton, M.J.; Long, F.; Ponder, K.P. Mechanism of shortened bones in mucopolysaccharidosis VII. Mol. Genet. Metab. 2009, 97, 202–211. [Google Scholar] [CrossRef]
- Jiang, Z.; Derrick-Roberts, A.L.K.; Jackson, M.R.; Rossouw, C.; Pyragius, C.E.; Xian, C.; Fletcher, J.; Byers, S. Delayed development of ossification centers in the tibia of prenatal and early postnatal MPS VII mice. Mol. Genet. Metab. 2018, 124, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Peck, S.H.; O’Donnell, P.J.M.; Kang, J.L.; Malhotra, N.R.; Dodge, G.R.; Pacifici, M.; Shore, E.M.; Haskins, M.E.; Smith, L.J. Delayed hypertrophic differentiation of epiphyseal chondrocytes contributes to failed secondary ossification in mucopolysaccharidosis VII dogs. Mol. Genet. Metab. 2015, 116, 195–203. [Google Scholar] [CrossRef]
- Peck, S.H.; Tobias, J.W.; Shore, E.M.; Malhotra, N.R.; Haskins, M.E.; Casal, M.L.; Smith, L.J. Molecular profiling of failed endochondral ossification in mucopolysaccharidosis VII. Bone 2019, 128, 115042. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, M.J.G.; Balbín, M.; López, J.M.; Alvarez, J.; Komori, T.; López-Otín, C. Collagenase 3 Is a Target of Cbfa1, a Transcription Factor of the runt Gene Family Involved in Bone Formation. Mol. Cell. Biol. 1999, 19, 4431–4442. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Hashamiyan, S.; Clarke, L.; Saftig, P.; Mort, J.; Dejica, V.M.; Brömme, D. Glycosaminoglycan-Mediated Loss of Cathepsin K Collagenolytic Activity in MPS I Contributes to Osteoclast and Growth Plate Abnormalities. Am. J. Pathol. 2009, 175, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter Syndromes: Mutual Correction of the Defect in Cultured Fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Transplant Outcomes in Mucopolysaccharidoses. Semin. Hematol. 2010, 47, 59–69. [Google Scholar] [CrossRef]
- Hofmann, A.; Heyde, C.-E.; Völker, A.; Schumann, E.; von der Höh, N.H. Treatment of Severe Kyphoscoliosis in Children with Mucopolysaccharidosis Type I (Pfaundler–Hurler Syndrome) Using the Growing Rod Technique: A Case Series with Mid-Term Results. World Neurosurg. 2020, 139, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.M.; Darowski, M.; Morris, P.; Wraith, J.E. Anaesthesia and mucopolysaccharidoses: A Review of Airway Problems in Children. Anaesthesia 1994, 49, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.M.; Sprung, J.; Weingarten, T.N.; Warner, M.E. Anesthesia for patients with mucopolysaccharidoses: Comprehensive review of the literature with emphasis on airway management. Bosn. J. Basic Med. Sci. 2018, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Park, C.; Maeng, S. Mucopolysaccharidoses I and II: Brief Review of Therapeutic Options and Supportive/Palliative Therapies. BioMed Res. Int. 2020, 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Otman, S.; Kose, N.; Yakut, Y. The efficacy of Schroth`s 3-dimensional exercise therapy in the treatment of adolescent idiopathic scoliosis in Turkey. Neurosciences 2005, 10, 277–283. [Google Scholar] [PubMed]
- Fusco, C.; Zaina, F.; Atanasio, S.; Romano, M.; Negrini, A.; Negrini, S. Physical exercises in the treatment of adolescent idiopathic scoliosis: An updated systematic review. Physiother. Theory Pract. 2011, 27, 80–114. [Google Scholar] [CrossRef] [PubMed]
- Marinela, R. Early Physical Therapy Intervention in Infant Hip Dysplasia. Procedia-Soc. Behav. Sci. 2013, 76, 729–733. [Google Scholar] [CrossRef][Green Version]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; DeFor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013, 121, 3981–3987. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic Cell Transplantation for Mucopolysaccharidosis Patients Is Safe and Effective: Results after Implementation of International Guidelines. Biol. Blood Marrow Transplant. 2015, 21, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Carbajal-Rodríguez, L.M.; Pérez-García, M.; Rodríguez-Herrera, R.; Rosales, H.S.; Olaya-Vargas, A. Long-term evolution of mucopolysaccharidosis type I in twins treated with enzyme replacement therapy plus hematopoietic stem cells transplantation. Heliyon 2021, 7, e07740. [Google Scholar] [CrossRef] [PubMed]
- Staba, S.L.; Escolar, M.L.; Poe, M.; Kim, Y.; Martin, P.L.; Szabolcs, P.; Allison-Thacker, J.; Wood, S.; Wenger, D.A.; Rubinstein, P.; et al. Cord-Blood Transplants from Unrelated Donors in Patients with Hurler’s Syndrome. N. Engl. J. Med. 2004, 350, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved Metabolic Correction in Patients with Lysosomal Storage Disease Treated with Hematopoietic Stem Cell Transplant Compared with Enzyme Replacement Therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Souillet, G.; Guffon, N.; Maire, I.; Pujol, M.; Taylor, P.; Sevin, F.; Bleyzac, N.; Mulier, C.; Durin, A.; Kebaili, K.; et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003, 31, 1105–1117. [Google Scholar] [CrossRef]
- Kunin-Batson, A.S.; Shapiro, E.G.; Rudser, K.D.; Lavery, C.A.; Bjoraker, K.J.; Jones, S.A.; Wynn, R.F.; Vellodi, A.; Tolar, J.; Orchard, P.J.; et al. Long-Term Cognitive and Functional Outcomes in Children with Mucopolysaccharidosis (MPS)-IH (Hurler Syndrome) Treated with Hematopoietic Cell Transplantation. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 29, pp. 95–102. ISBN 978-3-662-53277-5. [Google Scholar]
- Coletti, H.Y.; Aldenhoven, M.; Yelin, K.; Poe, M.D.; Kurtzberg, J.; Escolar, M.L. Long-Term Functional Outcomes of Children with Hurler Syndrome Treated with Unrelated Umbilical Cord Blood Transplantation. In JIMD Reports; Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 20, pp. 77–86. ISBN 978-3-662-46699-5. [Google Scholar]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in mucopolysaccharidosis type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef]
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early treatment is associated with improved cognition in Hurler syndrome: Early UCBT in MPS1. Ann. Neurol. 2014, 76, 747–753. [Google Scholar] [CrossRef]
- Vellodi, A.; Young, E.P.; Cooper, A.; Wraith, J.E.; Winchester, B.; Meaney, C.; Ramaswami, U.; Will, A. Bone marrow transplantation for mucopolysaccharidosis type I: Experience of two British centres. Arch. Dis. Child. 1997, 76, 92–99. [Google Scholar] [CrossRef]
- Yasuda, E.; Mackenzie, W.G.; Ruhnke, K.D.; Shimada, T.; Mason, R.W.; Zustin, J.; Martin, P.L.; Thacker, M.M.; Orii, T.; Sai, Y.; et al. Long-term follow-up of post hematopoietic stem cell transplantation for Hurler syndrome: Clinical, biochemical, and pathological improvements. Mol. Genet. Metab. Rep. 2015, 2, 65–76. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Rudser, K.D.; Tolar, J.; Orchard, P.J.; Kivisto, T.; Ziegler, R.S.; Whitley, C.B.; Shapiro, E.G. Enzyme Replacement is Associated with Better Cognitive Outcomes after Transplant in Hurler Syndrome. J. Pediatr. 2013, 162, 375–380.e1. [Google Scholar] [CrossRef]
- Field, R.E.; Buchanan, J.A.; Copplemans, M.G.; Aichroth, P.M. Bone-Marrow Transplantation in Hurler’s Syndrome. Effect on Skeletal Development. J. Bone Joint Surg. Br. 1994, 76, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Langereis, E.J.; den Os, M.M.; Breen, C.; Jones, S.A.; Knaven, O.C.; Mercer, J.; Miller, W.P.; Kelly, P.M.; Kennedy, J.; Ketterl, T.G.; et al. Progression of Hip Dysplasia in Mucopolysaccharidosis Type I Hurler After Successful Hematopoietic Stem Cell Transplantation. J. Bone Jt. Surg. 2016, 98, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Weisstein, J.S.; Delgado, E.; Steinbach, L.S.; Hart, K.; Packman, S. Musculoskeletal Manifestations of Hurler Syndrome. J. Pediatr. Orthop. 2004, 24, 5. [Google Scholar] [CrossRef]
- Schmidt, M.; Breyer, S.; Löbel, U.; Yarar, S.; Stücker, R.; Ullrich, K.; Müller, I.; Muschol, N. Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J. Rare Dis. 2016, 11, 93. [Google Scholar] [CrossRef]
- Odunusi, E.; Peters, C.; Krivit, W.; Ogilvie, J. Genu Valgum Deformity in Hurler Syndrome After Hematopoietic Stem Cell Transplantation: Correction by Surgical Intervention. J. Pediatr. Orthop. 1999, 19, 270–274. [Google Scholar] [CrossRef]
- Cooper, G.A.; Southorn, T.; Eastwood, D.M.; Bache, C.E. Lower Extremity Deformity Management in MPS IVA, Morquio-Brailsford Syndrome: Preliminary Report of Hemiepiphysiodesis Correction of Genu Valgum. J. Pediatr. Orthop. 2016, 36, 376–381. [Google Scholar] [CrossRef]
- Borgo, A.; Cossio, A.; Gallone, D.; Vittoria, F.; Carbone, M. Orthopaedic challenges for mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 123. [Google Scholar] [CrossRef]
- Pauchard, N.; Garin, C.; Jouve, J.L.; Lascombes, P.; Journeau, P. Perioperative Medullary Complications in Spinal and Extra-Spinal Surgery in Mucopolysaccharidosis: A Case Series of Three Patients. In JIMD Reports; Zschocke, J., Gibson, K.M., Brown, G., Morava, E., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 16, pp. 95–99. ISBN 978-3-662-44586-0. [Google Scholar]
- Cattoni, A.; Chiaraluce, S.; Gasperini, S.; Molinari, S.; Biondi, A.; Rovelli, A.; Parini, R. “Growth patterns in children with mucopolysaccharidosis type I-Hurler after hematopoietic stem cell transplantation: Comparison with untreated patients”. Mol. Genet. Metab. Rep. 2021, 28, 100787. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Thomas, W.; Orchard, P.J.; Whitley, C.B.; Miller, B.S. Effect of recombinant human growth hormone on changes in height, bone mineral density, and body composition over 1–2years in children with Hurler or Hunter syndrome. Mol. Genet. Metab. 2014, 111, 101–106. [Google Scholar] [CrossRef]
- Bakker, B.; Oostdijk, W.; Bresters, D.; Walenkamp, M.J.E.; Vossen, J.M.; Wit, J.M. Disturbances of growth and endocrine function after busulphan-based conditioning for haematopoietic stem cell transplantation during infancy and childhood. Bone Marrow Transplant. 2004, 33, 1049–1056. [Google Scholar] [CrossRef]
- Giorgiani, G.; Bozzola, M.; Locatelli, F.; Picco, P.; Zecca, M.; Cisternino, M.; Dallorso, S.; Bonetti, F.; Dini, G.; Borrone, C. Role of busulfan and total body irradiation on growth of prepubertal children receiving bone marrow transplantation and results of treatment with recombinant human growth hormone. Blood 1995, 86, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Cattoni, A.; Motta, S.; Masera, N.; Gasperini, S.; Rovelli, A.; Parini, R. The use of recombinant human growth hormone in patients with Mucopolysaccharidoses and growth hormone deficiency: A case series. Ital. J. Pediatr. 2019, 45, 93. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Zivicnjak, M.; Grigull, L.; Hennermann, J.B.; Aries, C.; Maecker-Kolhoff, B.; Sauer, M.; Das, A.M.; Beier, R. Predictors of growth patterns in children with mucopolysaccharidosis I after haematopoietic stem cell transplantation. JIMD Rep. 2022, 63, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.J.; Robinson, N.; Meadows, T.; Wynn, R.; Will, A.; Mercer, J.; Church, H.J.; Tylee, K.; Wraith, J.E.; Clayton, P.E. Growth, final height and endocrine sequelae in a UK population of patients with Hurler syndrome (MPS1H). J. Inherit. Metab. Dis. 2011, 34, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. [Google Scholar] [CrossRef]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-Replacement Therapy in Mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef]
- Dierenfeld, A.D.; McEntee, M.F.; Vogler, C.A.; Vite, C.H.; Chen, A.H.; Passage, M.; Le, S.; Shah, S.; Jens, J.K.; Snella, E.M.; et al. Replacing the Enzyme α- l -Iduronidase at Birth Ameliorates Symptoms in the Brain and Periphery of Dogs with Mucopolysaccharidosis Type I. Sci. Transl. Med. 2010, 2, 60ra89. [Google Scholar] [CrossRef]
- Gabrielli, O.; Clarke, L.A.; Ficcadenti, A.; Santoro, L.; Zampini, L.; Volpi, N.; Coppa, G.V. 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: The important role of early treatment. BMC Med. Genet. 2016, 17, 19. [Google Scholar] [CrossRef]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Guelbert, N.; Jones, S.A.; Kyosen, S.O.; Martins, A.M.; Phornphutkul, C.; et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: A retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 131. [Google Scholar] [CrossRef]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-L-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef]
- Pitz, S.; Ogun, O.; Bajbouj, M.; Arash, L.; Schulze-Frenking, G.; Beck, M. Ocular Changes in Patients with Mucopolysaccharidosis I Receiving Enzyme Replacement Therapy: A 4-Year Experience. Arch. Ophthalmol. 2007, 125, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Laraway, S.; Mercer, J.; Jameson, E.; Ashworth, J.; Hensman, P.; Jones, S.A. Outcomes of Long-Term Treatment with Laronidase in Patients with Mucopolysaccharidosis Type I. J. Pediatr. 2016, 178, 219–226.e1. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymańska, A.; Marucha, J.; Jurecka, A.; Syczewska, M.; Czartoryska, B. Efficacy of recombinant human α-L-iduronidase (laronidase) on restricted range of motion of upper extremities in mucopolysaccharidosis type I patients. J. Inherit. Metab. Dis. 2010, 33, 151–157. [Google Scholar] [CrossRef]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- D’Aco, K.; Underhill, L.; Rangachari, L.; Arn, P.; Cox, G.F.; Giugliani, R.; Okuyama, T.; Wijburg, F.; Kaplan, P. Diagnosis and treatment trends in mucopolysaccharidosis I: Findings from the MPS I Registry. Eur. J. Pediatr. 2012, 171, 911–919. [Google Scholar] [CrossRef]
- Franco, J.F.; Soares, D.C.; Torres, L.C.; Leal, G.N.; Cunha, M.T.; Honjo, R.S.; Bertola, D.R.; Kim, C.A. Short Communication Impact of early enzyme-replacement therapy for mucopolysaccharidosis VI: Results of a long-term follow-up of Brazilian siblings. Genet. Mol. Res. 2016, 15, 1–7. [Google Scholar] [CrossRef]
- McGill, J.; Inwood, A.; Coman, D.; Lipke, M.; De Lore, D.; Swiedler, S.; Hopwood, J. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age–A sibling control study. Clin. Genet. 2010, 77, 492–498. [Google Scholar] [CrossRef]
- Baldo, G.; Mayer, F.Q.; Martinelli, B.Z.; de Carvalho, T.G.; Meyer, F.S.; de Oliveira, P.G.; Meurer, L.; Tavares, Â.; Matte, U.; Giugliani, R. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2013, 109, 33–40. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.-W.; Lu, J.Z.; Zhou, Q.-H.; Pardridge, W.M. Reversal of Lysosomal Storage in Brain of Adult MPS-I Mice with Intravenous Trojan Horse-Iduronidase Fusion Protein. Mol. Pharm. 2011, 8, 1342–1350. [Google Scholar] [CrossRef]
- Giugliani, R.; Giugliani, L.; de Oliveira Poswar, F.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): An open label phase 1-2 trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef]
- Grewal, S.S.; Wynn, R.; Abdenur, J.E.; Burton, B.K.; Gharib, M.; Haase, C.; Hayashi, R.J.; Shenoy, S.; Sillence, D.; Tiller, G.E.; et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet. Med. 2005, 7, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Miller, W.; Orchard, P.J.; Jones, S.A.; Mercer, J.; Church, H.J.; Tylee, K.; Lund, T.; Bigger, B.W.; Tolar, J.; et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in Mucopolysaccharidosis Type I: 10year combined experience of 2 centres. Mol. Genet. Metab. 2016, 117, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; de Blic, J.; Mahlaoui, N.; Stos, B.; Jaubert, F.; Bonnet, D.; Fischer, A.; de Lonlay, P. Laronidase for Cardiopulmonary Disease in Hurler Syndrome 12 Years After Bone Marrow Transplantation. Pediatrics 2010, 126, e1242–e1247. [Google Scholar] [CrossRef] [PubMed]
- Tolar, J.; Grewal, S.S.; Bjoraker, K.J.; Whitley, C.B.; Shapiro, E.G.; Charnas, L.; Orchard, P.J. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008, 41, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Matte, U.D.S.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.A.; Bigger, B.W.; Brookes, K.E.; Mercer, J.; Tylee, K.L.; Church, H.J.; Bonney, D.K.; Jones, S.; Wraith, J.E.; Wynn, R.F. Hematopoietic stem cell transplantation improves the high incidence of neutralizing allo-antibodies observed in Hurler’s syndrome after pharmacological enzyme replacement therapy. Haematologica 2012, 97, 1320–1328. [Google Scholar] [CrossRef]
- Ferrara, G.; Maximova, N.; Zennaro, F.; Gregori, M.; Tamaro, P. Hematopoietic stem cell transplantation effects on spinal cord compression in Hurler. Pediatr. Transplant. 2014, 18, E96–E99. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.R.; Langereis, E.J.; Saif, M.A.; Mercer, J.; Church, H.J.; Tylee, K.L.; Wynn, R.F.; Wijburg, F.A.; Jones, S.A.; Bruce, I.A.; et al. Sleep disordered breathing in mucopolysaccharidosis I: A multivariate analysis of patient, therapeutic and metabolic correlators modifying long term clinical outcome. Orphanet J. Rare Dis. 2015, 10, 42. [Google Scholar] [CrossRef][Green Version]
- Connock, M.; Juarez-Garcia, A.; Frew, E.; Mans, A.; Dretzke, J.; Fry-Smith, A.; Moore, D. A systematic review of the clinical effectiveness and cost-effectiveness of enzyme replacement therapies for Fabry’s disease and mucopolysaccharidosis type 1. Health Technol. Assess. 2006, 10, iii–iv. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Tittiger, M.; Hennig, A.; Kovacs, A.; Popelka, S.; Wang, B.; Herati, R.; Bigg, M.; Ponder, K.P. Improvements in Mucopolysaccharidosis I Mice After Adult Retroviral Vector–mediated Gene Therapy with Immunomodulation. Mol. Ther. 2007, 15, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Carbonaro, D.; Pepper, K.; Petersen, D.; Ge, S.; Jackson, H.; Shimada, H.; Moats, R.; Kohn, D.B. Neonatal Gene Therapy of MPS I Mice by Intravenous Injection of a Lentiviral Vector. Mol. Ther. 2005, 11, 776–789. [Google Scholar] [CrossRef]
- Mango, R.L. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol. Genet. Metab. 2004, 82, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Hartung, S.D.; Frandsen, J.L.; Pan, D.; Koniar, B.L.; Graupman, P.; Gunther, R.; Low, W.C.; Whitley, C.B.; McIvor, R.S. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human α-l-iduronidase gene. Mol. Ther. 2004, 9, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Barrera, L.A.; Tomatsu, S. Tailoring the AAV2 capsid vector for bone-targeting. Pediatr. Res. 2018, 84, 545–551. [Google Scholar] [CrossRef]
- Taylor, R.M.; Wolfe, O.H. Decreased Lysosomal Storage in the Adult MPS VII Mouse Brain in the Vicinity of Grafts of Retroviral Vector-Corrected Fibroblasts Secreting High Levels of B-Glucuronidase. Nat. Med. 1997, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-M.; Wong, A.; Yu, X.; Kakkis, E.; Kohn, D. Retrovirus-mediated transfer of the human α- L-iduronidase cDNA into human hematopoietic progenitor cells leads to correction in trans of Hurler fibroblasts. Gene Ther. 1997, 4, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.A.; Wynn, R.F.; Deakin, J.A.; Bellantuono, I.; Edington, K.G.; Cooper, A.; Besley, G.T.N.; Church, H.J.; Wraith, J.E.; Carr, T.F.; et al. Retrovirally mediated correction of bone marrow–derived mesenchymal stem cells from patients with mucopolysaccharidosis type I. Blood 2002, 99, 1857–1859. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, C.; Di Napoli, D.; Reyero, E.G.Y.; Lombardo, A.; Naldini, L.; Di Natale, P. Limited Transgene Immune Response and Long-Term Expression of Humanα-L-Iduronidase in Young Adult Mice with Mucopolysaccharidosis Type I by Liver-Directed Gene Therapy. Hum. Gene Ther. 2006, 17, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Traas, A.M.; Wang, P.; Ma, X.; Tittiger, M.; Schaller, L.; O’Donnell, P.; Sleeper, M.M.; Vite, C.; Herati, R.; Aguirre, G.D.; et al. Correction of Clinical Manifestations of Canine Mucopolysaccharidosis I with Neonatal Retroviral Vector Gene Therapy. Mol. Ther. 2007, 15, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, C.; Villani, G.R.D.; Di Napoli, D.; Reyero, E.G.Y.; Lombardo, A.L.; Naldini, L.; Di Natale, P. Gene Therapy for a Mucopolysaccharidosis Type I Murine Model with Lentiviral-IDUA Vector. Hum. Gene Ther. 2005, 16, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Scala, S.; Basso-Ricci, L.; Dionisio, F.; Pellin, D.; Giannelli, S.; Salerio, F.A.; Leonardelli, L.; Cicalese, M.P.; Ferrua, F.; Aiuti, A.; et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat. Med. 2018, 24, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Rozengurt, N.; Ryazantsev, S.; Kohn, D.B.; Satake, N.; Neufeld, E.F. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol. Genet. Metab. 2003, 79, 233–244. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, W.; Kalfa, T.A.; Grabowski, G.; Davies, S.; Malik, P.; Pan, D. Reprogramming erythroid cells for lysosomal enzyme production leads to visceral and CNS cross-correction in mice with Hurler syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 19958–19963. [Google Scholar] [CrossRef] [PubMed]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Visigalli, D.I.; Delai, D.S.; Ferro, D.F.; Cecere, D.F.; Vezzoli, D.M.; Sanvito, D.F.; Chanut, D.F.; Benedicenti, D.F.; Spinozzi, D.G.; Wynn, R.; et al. Preclinical Testing of the Safety and Tolerability of LV-Mediated above Normal Alpha-L-Iduronidase Expression in Murine and Human Hematopoietic Cells Using Toxicology and Biodistribution GLP Studies. Hum. Gene Ther. 2016, 27, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; De Pellegrin, M.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Weinberg, K.I.; Nolta, J.A.; Heiss, L.N.; Lenarsky, C.; Crooks, G.M.; Hanley, M.E.; Annett, G.; Brooks, J.S.; El-Khoureiy, A.; et al. Engraftment of gene–modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat. Med. 1995, 1, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome Engineering with Targetable Nucleases. Annu. Rev. Biochem. 2014, 83, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Poletto, E.; Pasqualim, G.; Tavares, A.M.V.; Meyer, F.S.; Gonzalez, E.A.; Giugliani, R.; Matte, U.; Teixeira, H.F.; Baldo, G. In vivo genome editing of mucopolysaccharidosis I mice using the CRISPR/Cas9 system. J. Control. Release 2018, 288, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.; Lau, H.A.; Heldermon, C.; Leslie, N.; Foo, C.W.P.; Vaidya, S.A.; Whitley, C.B. EMPOWERS: A phase 1/2 clinical trial of SB-318 ZFN-mediated in vivo human genome editing for treatment of MPS I (Hurler syndrome). Mol. Genet. Metab. 2019, 126, S68. [Google Scholar] [CrossRef]
- Aronovich, E.L.; Bell, J.B.; Khan, S.A.; Belur, L.R.; Gunther, R.; Koniar, B.; Schachern, P.A.; Parker, J.B.; Carlson, C.S.; Whitley, C.B.; et al. Systemic Correction of Storage Disease in MPS I NOD/SCID Mice Using the Sleeping Beauty Transposon System. Mol. Ther. 2009, 17, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Sena-Esteves, M.; Gao, G. Introducing Genes into Mammalian Cells: Viral Vectors. Cold Spring Harb. Protoc. 2020, 2020, pdb-top095513. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Beck, M. New therapeutic options for lysosomal storage disorders: Enzyme replacement, small molecules and gene therapy. Hum. Genet. 2006, 121, 1–22. [Google Scholar] [CrossRef]
- Jakobkiewicz-Banecka, J.; Piotrowska, E.; Gabig-Cimińska, M.; Borysiewicz, E.; Słomińska-Wojewódzka, M.; Narajczyk, M.; Wegrzyn, A.; Wegrzyn, G. Substrate reduction therapies for mucopolysaccharidoses. Curr. Pharm. Biotechnol. 2011, 12, 1860–1865. [Google Scholar] [CrossRef]
- Jakóbkiewicz-Banecka, J.; Piotrowska, E.; Narajczyk, M.; Barańska, S.; Węgrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 2009, 16, 26. [Google Scholar] [CrossRef]
- Marucha, J.; Tylki-Szymańska, A.; Jakóbkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Węgrzyn, G. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am. J. Med. Genet. Part A 2011, 155, 2257–2262. [Google Scholar] [CrossRef]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Maryniak, A.; Tylki-Szymanska, A.; Puk, E.; Liberek, A.; Wegrzyn, A.; Czartoryska, B.; Slominska-Wojewodzka, M.; Wegrzyn, G. Two-year follow-up of Sanfilippo Disease patients treated with a genistein-rich isoflavone extract: Assessment of effects on cognitive functions and general status of patients. Med Sci. Monit. 2011, 17, CR196–CR202. [Google Scholar] [CrossRef]
- Kingma, S.D.K.; Wagemans, T.; IJlst, L.; Seppen, J.; Gijbels, M.J.J.; Wijburg, F.A.; van Vlies, N. Adverse Effects of Genistein in a Mucopolysaccharidosis Type I Mouse Model. In JIMD Reports; Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 23, pp. 77–83. ISBN 978-3-662-47466-2. [Google Scholar]
- Kaidonis, X.; Byers, S.; Ranieri, E.; Sharp, P.; Fletcher, J.; Derrick-Roberts, A. N-butyldeoxynojirimycin treatment restores the innate fear response and improves learning in mucopolysaccharidosis IIIA mice. Mol. Genet. Metab. 2016, 118, 100–110. [Google Scholar] [CrossRef]
- Kaidonis, X.; Liaw, W.C.; Roberts, A.D.; Ly, M.; Anson, D.; Byers, S. Gene silencing of EXTL2 and EXTL3 as a substrate deprivation therapy for heparan sulphate storing mucopolysaccharidoses. Eur. J. Hum. Genet. 2010, 18, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of Nonsense-Mediated mRNA Decay Enhances In vivo Nonsense Suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [PubMed]
- Gunn, G.; Dai, Y.; Du, M.; Belakhov, V.; Kandasamy, J.; Schoeb, T.R.; Baasov, T.; Bedwell, D.M.; Keeling, K.M. Long-term nonsense suppression therapy moderates MPS I-H disease progression. Mol. Genet. Metab. 2014, 111, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and Characterization of Pharmacological Chaperones to Correct Enzyme Deficiencies in Lysosomal Storage Disorders. ASSAY Drug Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.K.; Kakkis, E.D. The multi-domain responder index: A novel analysis tool to capture a broader assessment of clinical benefit in heterogeneous complex rare diseases. Orphanet J. Rare Dis. 2021, 16, 183. [Google Scholar] [CrossRef]
- Available online: https://Clinicaltrials.Gov/Ct2/Show/Results/NCT00146757?View=results (accessed on 16 September 2022).
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef]
- Arn, P.; Bruce, I.A.; Wraith, J.E.; Travers, H.; Fallet, S. Airway-related symptoms and surgeries in patients with mucopolysaccharidosis I. Ann. Otol. Rhinol. Laryngol. 2015, 124, 198–205. [Google Scholar] [CrossRef]
- Available online: https://Clinicaltrials.Gov/Ct2/Show/Results/NCT00144781?Cond=NCT00144781&draw=2&rank=1 (accessed on 16 September 2022).
- Castorina, M.; Antuzzi, D.; Richards, S.M.; Cox, G.F.; Xue, Y. Successful pregnancy and breastfeeding in a woman with mucopolysaccharidosis type I while receiving laronidase enzyme replacement therapy. Clin. Exp. Obstet. Gynecol. 2015, 42, 108–113. [Google Scholar] [CrossRef]
- Available online: https://Clinicaltrials.Gov/Ct2/Show/Results/NCT01173016?Cond=NCT01173016&draw=2&rank=1 (accessed on 16 September 2022).
- Available online: https://Clinicaltrials.Gov/Ct2/Show/Results/NCT02437253?Cond=NCT02437253&draw=2&rank=1 (accessed on 16 September 2022).
- Simonaro, C.M.; Tomatsu, S.; Sikora, T.; Kubaski, F.; Frohbergh, M.; Guevara, J.M.; Wang, R.Y.; Vera, M.; Kang, J.L.; Smith, L.J.; et al. Pentosan Polysulfate: Oral Versus Subcutaneous Injection in Mucopolysaccharidosis Type I Dogs. PLoS ONE 2016, 11, e0153136. [Google Scholar] [CrossRef] [PubMed]
- Hennermann, J.B.; Gökce, S.; Solyom, A.; Mengel, E.; Schuchman, E.H.; Simonaro, C.M. Treatment with pentosan polysulphate in patients with MPS I: Results from an open label, randomized, monocentric phase II study. J. Inherit. Metab. Dis. 2016, 39, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Eliyahu, E.; Wolfson, T.; Ge, Y.; Jepsen, K.J.; Schuchman, E.H.; Simonaro, C.M. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE 2011, 6, e22447. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Fujii, T.; Yamauchi, I.; Ueda, Y.; Hirota, K.; Kanai, Y.; Yasoda, A.; Inagaki, N. C-Type Natriuretic Peptide Restores Growth Impairment Under Enzyme Replacement in Mice with Mucopolysaccharidosis VII. Endocrinology 2020, 161, bqaa008. [Google Scholar] [CrossRef]
- Bartolomeo, R.; Cinque, L.; De Leonibus, C.; Forrester, A.; Salzano, A.C.; Monfregola, J.; De Gennaro, E.; Nusco, E.; Azario, I.; Lanzara, C.; et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Investig. 2017, 127, 3717–3729. [Google Scholar] [CrossRef]
- Nan, Z.; Shekels, L.; Ryabinin, O.; Evavold, C.; Nelson, M.S.; Khan, S.A.; Deans, R.J.; Mays, R.W.; Low, W.C.; Gupta, P. Intracerebroventricular Transplantation of Human Bone Marrow-Derived Multipotent Progenitor Cells in an Immunodeficient Mouse Model of Mucopolysaccharidosis Type I (MPS-I). Cell Transplant. 2012, 21, 1577–1593. [Google Scholar] [CrossRef]
- Meyerrose, T.E.; Roberts, M.; Ohlemiller, K.K.; Vogler, C.A.; Wirthlin, L.; Nolta, J.A.; Sands, M.S. Lentiviral-Transduced Human Mesenchymal Stem Cells Persistently Express Therapeutic Levels of Enzyme in a Xenotransplantation Model of Human Disease. Stem Cells 2008, 26, 1713–1722. [Google Scholar] [CrossRef][Green Version]
- Koç, O.; Day, J.; Nieder, M.; Gerson, S.; Lazarus, H.; Krivit, W. Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH). Bone Marrow Transplant. 2002, 30, 215–222. [Google Scholar] [CrossRef]
- Iolascon, G.; Moretti, A. The Rationale for Using Neridronate in Musculoskeletal Disorders: From Metabolic Bone Diseases to Musculoskeletal Pain. Int. J. Mol. Sci. 2022, 23, 6921. [Google Scholar] [CrossRef]
- Tummolo, A.; Gabrielli, O.; Gaeta, A.; Masciopinto, M.; Zampini, L.; Pavone, L.M.; Di Natale, P.; Papadia, F. Bisphosphonate Treatment in a Patient Affected by MPS IVA with Osteoporotic Phenotype. Case Rep. Med. 2013, 2013, 1–4. [Google Scholar] [CrossRef]
- Wang, R.Y.; Cambray-Forker, E.J.; Ohanian, K.; Karlin, D.S.; Covault, K.K.; Schwartz, P.H.; Abdenur, J.E. Treatment reduces or stabilizes brain imaging abnormalities in patients with MPS I and II. Mol. Genet. Metab. 2009, 98, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, N.J.; Kaizer, A.M.; Miller, W.P.; Rudser, K.D.; Orchard, P.J.; Braunlin, E.A. Mortality after hematopoietic stem cell transplantation for severe mucopolysaccharidosis type I: The 30-year University of Minnesota experience. J. Inherit. Metab. Dis. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Montaño, A.M.; Oikawa, H.; Dung, V.C.; Hashimoto, A.; Oguma, T.; Gutiérrez, M.L.; Takahashi, T.; Shimada, T.; Orii, T.; et al. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: Early treatment rescues bone lesions? Mol. Genet. Metab. 2015, 114, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Azario, I.; Pievani, A.; Del Priore, F.; Antolini, L.; Santi, L.; Corsi, A.; Cardinale, L.; Sawamoto, K.; Kubaski, F.; Gentner, B.; et al. Neonatal umbilical cord blood transplantation halts skeletal disease progression in the murine model of MPS-I. Sci. Rep. 2017, 7, 9473. [Google Scholar] [CrossRef] [PubMed]
- Pievani, A.; Azario, I.; Antolini, L.; Shimada, T.; Patel, P.; Remoli, C.; Rambaldi, B.; Valsecchi, M.G.; Riminucci, M.; Biondi, A.; et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 2015, 125, 1662–1671. [Google Scholar] [CrossRef]
- Santi, L.; De Ponti, G.; Dina, G.; Pievani, A.; Corsi, A.; Riminucci, M.; Khan, S.; Sawamoto, K.; Antolini, L.; Gregori, S.; et al. Neonatal combination therapy improves some of the clinical manifestations in the Mucopolysaccharidosis type I murine model. Mol. Genet. Metab. 2020, 130, 197–208. [Google Scholar] [CrossRef]
- Hinderer, C.; Bell, P.; Louboutin, J.-P.; Zhu, Y.; Yu, H.; Lin, G.; Choa, R.; Gurda, B.L.; Bagel, J.; O’Donnell, P.; et al. Neonatal Systemic AAV Induces Tolerance to CNS Gene Therapy in MPS I Dogs and Nonhuman Primates. Mol. Ther. 2015, 23, 1298–1307. [Google Scholar] [CrossRef]
- Meng, X.-L.; Shen, J.-S.; Ohashi, T.; Maeda, H.; Kim, S.U.; Eto, Y. Brain transplantation of genetically engineered human neural stem cells globally corrects brain lesions in the mucopolysaccharidosis type VII mouse. J. Neurosci. Res. 2003, 74, 266–277. [Google Scholar] [CrossRef]
- Bose, S.K.; White, B.M.; Kashyap, M.V.; Dave, A.; De Bie, F.R.; Li, H.; Singh, K.; Menon, P.; Wang, T.; Teerdhala, S.; et al. In utero adenine base editing corrects multi-organ pathology in a lethal lysosomal storage disease. Nat. Commun. 2021, 12, 4291. [Google Scholar] [CrossRef]
- Casal, M.L.; Wolfe, J.H. In utero transplantation of fetal liver cells in the mucopolysaccharidosis type VII mouse results in low-level chimerism, but overexpression of β-glucuronidase can delay onset of clinical signs. Blood 2001, 97, 1625–1634. [Google Scholar] [CrossRef]
- Barker, J.E.; Deveau, S.; Lessard, M.; Hamblen, N.; Vogler, C.; Levy, B. In Utero Fetal Liver Cell Transplantation without Toxic Irradiation Alleviates Lysosomal Storage in Mice with Mucopolysaccharidosis Type VII. Blood Cells Mol. Dis. 2001, 27, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef] [PubMed]





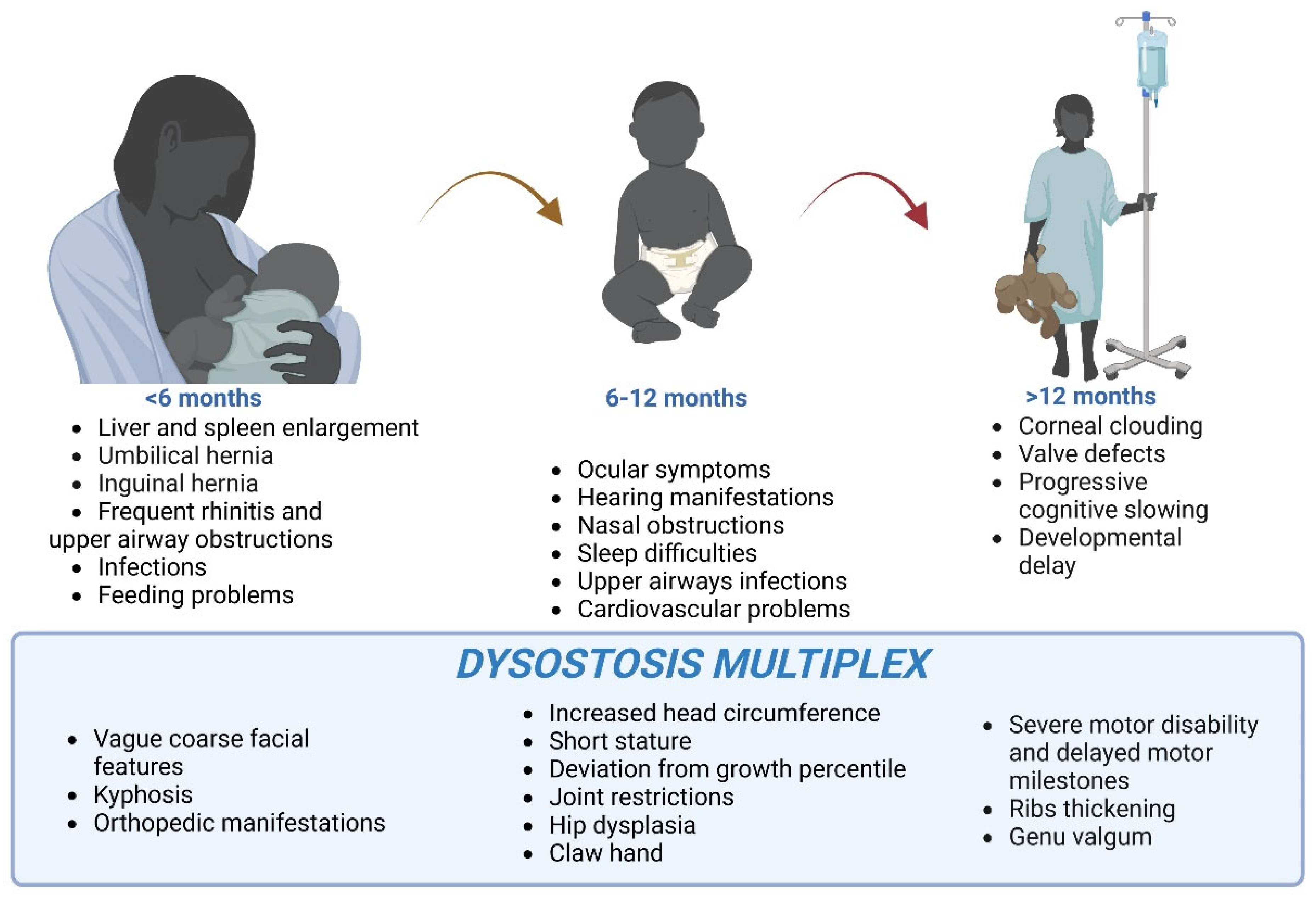{kind=link}
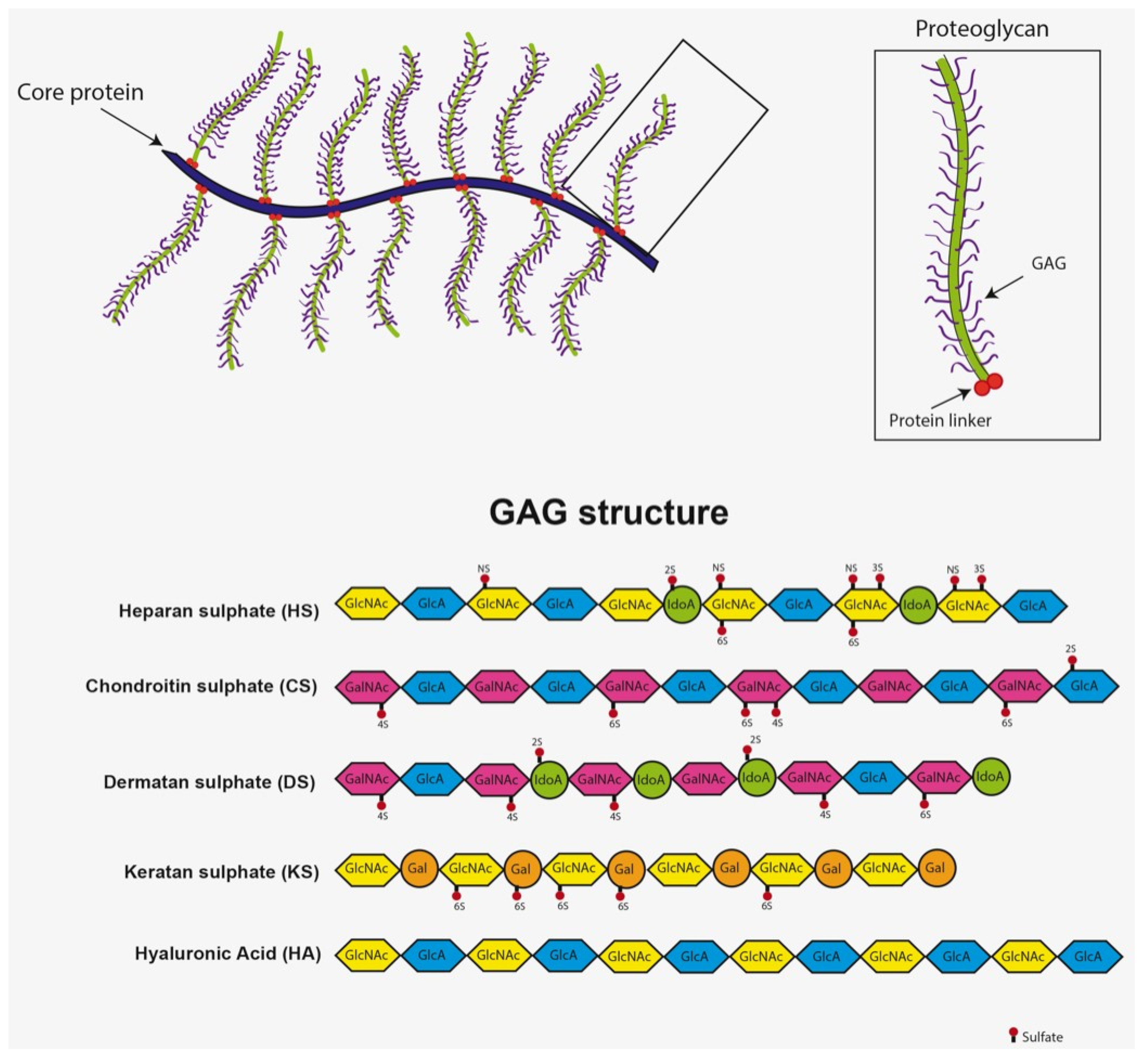{kind=link}
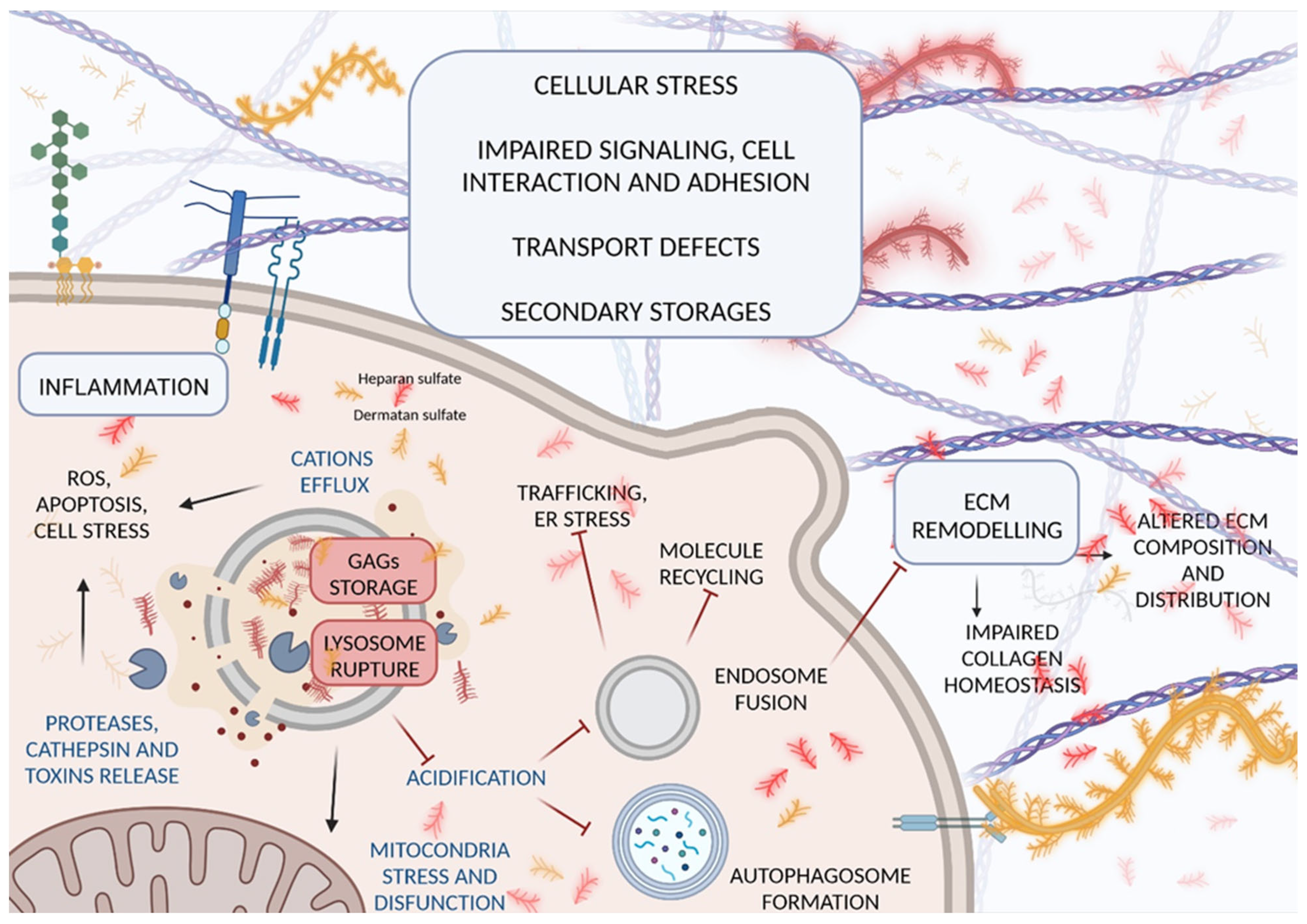{kind=link}
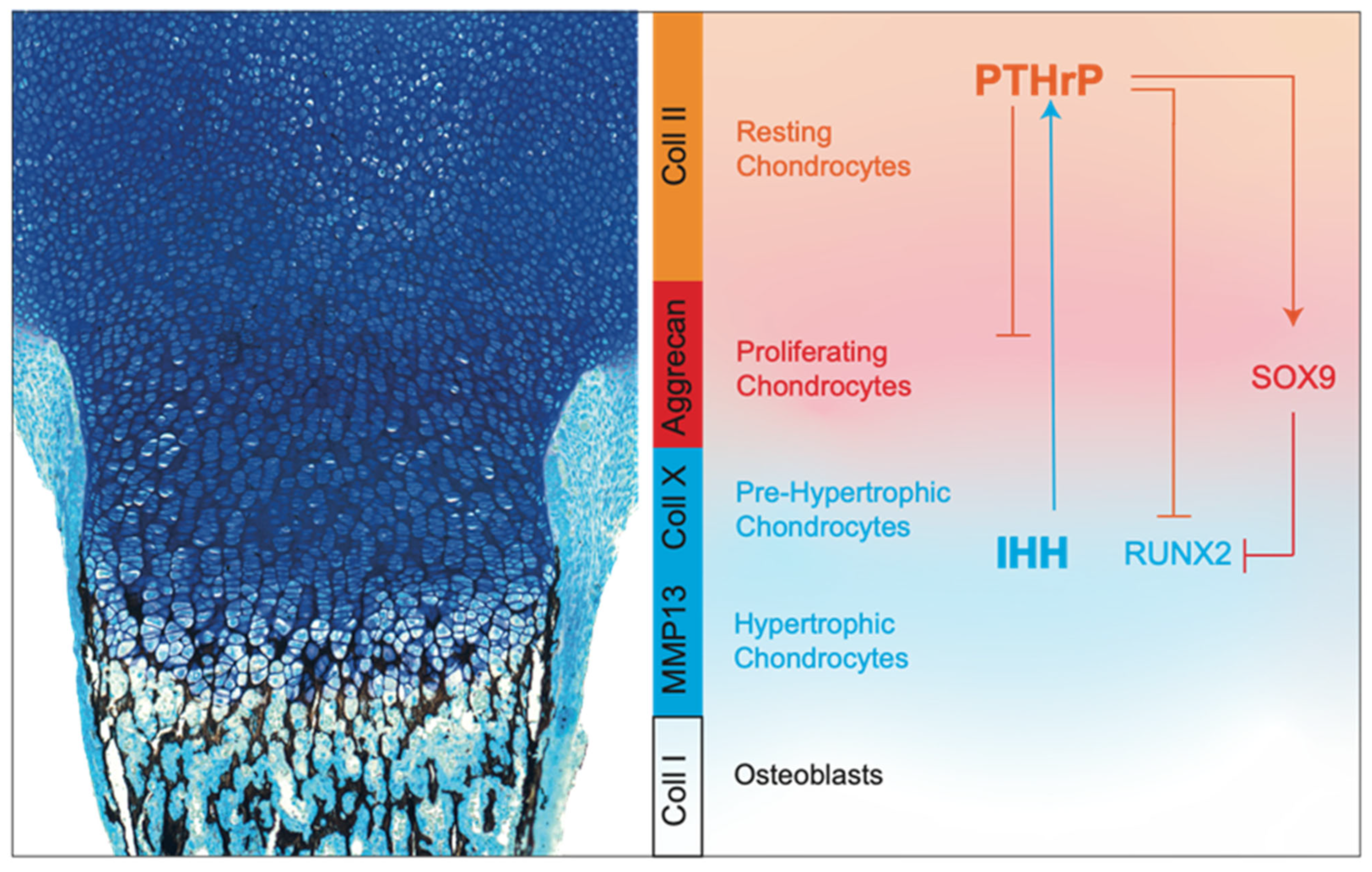{kind=link}
| Identifier (Phase/Type) | Date | Title | Sponsor Name | Age (Enrolled Patients) | Musculoskeletal Investigation | Results |
|---|---|---|---|---|---|---|
| NCT00912925 (3) | 2000–2001 | Clinical Study of Aldurazyme in Patients With Mucopolysaccharidosis (MPS) I | Genzyme (Sanofi Company) | ≥5 years (45) | 6-min walk test distance, active joint range of motion (+26 weeks) | [244] |
| NCT00146770 (3) | 2001–2005 | Phase 3 Extension Study of the Safety and Efficacy of Aldurazyme® (Laronidase) in Mucopolysaccharidosis I (MPS I) Patients | Genzyme (Sanofi Company) | Child, adult (45) | 6-min walk test distance, active joint range of motion (+182 weeks) | [244] |
| NCT00146757 (2) | 2002–2005 | A Study Evaluating the Safety and Pharmacokinetics of Aldurazyme® (Laronidase) in MPS I Patients Less Than 5 Years Old | Genzyme (Sanofi Company) | ≤5 years (20) | Measure of standing height/lying-length-for-age (+52 weeks) | [245] |
| NCT00144794 (observational) | 2003- | Mucopolysaccharidosis I (MPS I) Registry | Genzyme (Sanofi Company) | Child, adult (1500 *) | Description of variability, progression, and natural history of MPS I | [34,246,247] |
| NCT00144781 (4) | 2004–2006 | A Dose-optimization Study of Aldurazyme® (Laronidase) in Patients With Mucopolysaccharidosis I (MPS I) Disease | Genzyme (Sanofi Company) | Child, adult (34) | 6-min walk test distance (+26 weeks) | [248] |
| NCT01521429 (observational) | 2009–2019 | Longitudinal Study of Bone Disease in Children With Mucopolysaccharidoses (MPS) I, II, and VI | Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center | 5–35 years (55) | Analyses of bone density, geometry, strength and muscle fat, bone turnover, growth measurements (+1–2–3 years) | NA |
| NCT00418821 (4) | 2010- | A Study of the Effect of Aldurazyme® (Laronidase) Treatment on Lactation in Female Patients With Mucopolysaccharidosis I (MPS I) and Their Breastfed Infants | Genzyme (Sanofi Company) | Child, adult (2 *) | Drug effect in infants in terms of growth and development | [249] |
| NCT01173016 (1) | 2012–2016 | Administration of IV Laronidase Post Bone Marrow Transplant in Hurler | Masonic Cancer Center, University of Minnesota | 5–13 years (11) | Analyses in growth velocity, muscle strength, joint range of motion, 6-min walk test distance (+24 months) | [250] |
| NCT02067650 (observational) | 2013–2016 | Ultrasound Findings of Finger, Wrist and Knee Joints in Mucopolysaccharidosis | Children’s Hospital of Eastern Ontario | 2–99 years (18) | Finger, wrist, and knee joint abnormalities, synovitis/tenosynovitis | NA |
| NCT02171104 (2) | 2014- | MT2013-31: Allo-HCT for Metabolic Disorders and Severe Osteopetrosis | Masonic Cancer Center, University of Minnesota | ≤55 years (100 *) | Incidence of radiographic aspects of the disease (+1–2 years) | NA |
| NCT02437253 (1/2) | 2015–2017 | Effects of Adalimumab in Mucopolysaccharidosis Types I, II and VI | Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center | ≥5 years (2) | Height and weight, physical function, joint range of motion, 6-min walk test distance, and strength testing, serum joint inflammation (+16–32 weeks) | [251] |
| NCT03053089 (1/2) | 2015–2018 | Safety and Dose Ranging Study of Human Insulin Receptor MAb-IDUA Fusion Protein in Adults and Children With MPS I | ArmaGen, Inc. | ≥2 years (21) | Functional capacity in terms of 6-min walk test distance, shoulder range of motion (+26 weeks) | [196] |
| NCT03153319 (1/2) | 2017- | Study to Evaluate the Safety and Efficacy of Adalimumab in MPS I, II, and VI | Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center | ≥5 years (14 *) | Joint range of motion (+16–32 weeks) | NA |
| NCT02298712 (observational) | 2018- | Biomarker for Hurler Disease (BioHurler) | CENTOGENE GmbH Rostock | ≥2 months (1000 *) | PB markers for early diagnosis and avoidance of musculoskeletal manifestation appearance | NA |
| NCT03488394 (1/2) | 2018- | Gene Therapy With Modified Autologous Hematopoietic Stem Cells for the Treatment of Patients With Mucopolysaccharidosis Type I, Hurler Variant (TigetT10_MPSIH) | IRCCS San Raffaele | ≤11 years (8) | Growth, range of motion, clinical and radiological evaluations (+1–3–5 years) | [222] |
| NCT04227600 (1/2) | 2020- | A Study of JR-171 in Patients With Mucopolysaccharidosis I | JCR Pharmaceuticals Co., Ltd. | Child, adult (19 *) | 6-min walk test distance (+13 weeks) | NA |
| NCT04453085 (1/2) | 2021- | An Extension Study of JR-171-101 Study in Patients With MPS I | JCR Pharmaceuticals Co., Ltd. | Child, adult (15 *) | 6-min walk test distance | NA |
| NCT04958070 (observational) | 2021- | The Intensively Follow-up Examinations for Asymptomatic MPS I Infants in Taiwan | Mackay Memorial Hospital | 3–8 years (median, 16) | Regular physical examinations | NA |
| NCT04532047 (1) | 2021- | In Utero Enzyme Replacement Therapy for Lysosomal Storage Diseases (IUERT) | University of California, San Francisco | 18–50 years (10 *) | Growth, mobility, skeletal survey evaluations (+6 years) | NA |
| NCT04284254 (1/2) | 2022- | MT2018-18: Sleeping Beauty Transposon-Engineered Plasmablasts for Hurler Syndrome Post-Allo-HSCT | Masonic Cancer Center, University of Minnesota | 3–8 years (36 *) | Growth velocity in terms of cm/year, sitting and standing height (+1 year) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Ponti, G.; Donsante, S.; Frigeni, M.; Pievani, A.; Corsi, A.; Bernardo, M.E.; Riminucci, M.; Serafini, M. MPSI Manifestations and Treatment Outcome: Skeletal Focus. Int. J. Mol. Sci. 2022, 23, 11168. https://doi.org/10.3390/ijms231911168
De Ponti G, Donsante S, Frigeni M, Pievani A, Corsi A, Bernardo ME, Riminucci M, Serafini M. MPSI Manifestations and Treatment Outcome: Skeletal Focus. International Journal of Molecular Sciences. 2022; 23(19):11168. https://doi.org/10.3390/ijms231911168
Chicago/Turabian StyleDe Ponti, Giada, Samantha Donsante, Marta Frigeni, Alice Pievani, Alessandro Corsi, Maria Ester Bernardo, Mara Riminucci, and Marta Serafini. 2022. "MPSI Manifestations and Treatment Outcome: Skeletal Focus" International Journal of Molecular Sciences 23, no. 19: 11168. https://doi.org/10.3390/ijms231911168
APA StyleDe Ponti, G., Donsante, S., Frigeni, M., Pievani, A., Corsi, A., Bernardo, M. E., Riminucci, M., & Serafini, M. (2022). MPSI Manifestations and Treatment Outcome: Skeletal Focus. International Journal of Molecular Sciences, 23(19), 11168. https://doi.org/10.3390/ijms231911168