

Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration



Abstract
:1. Introduction
2. Results and Discussion
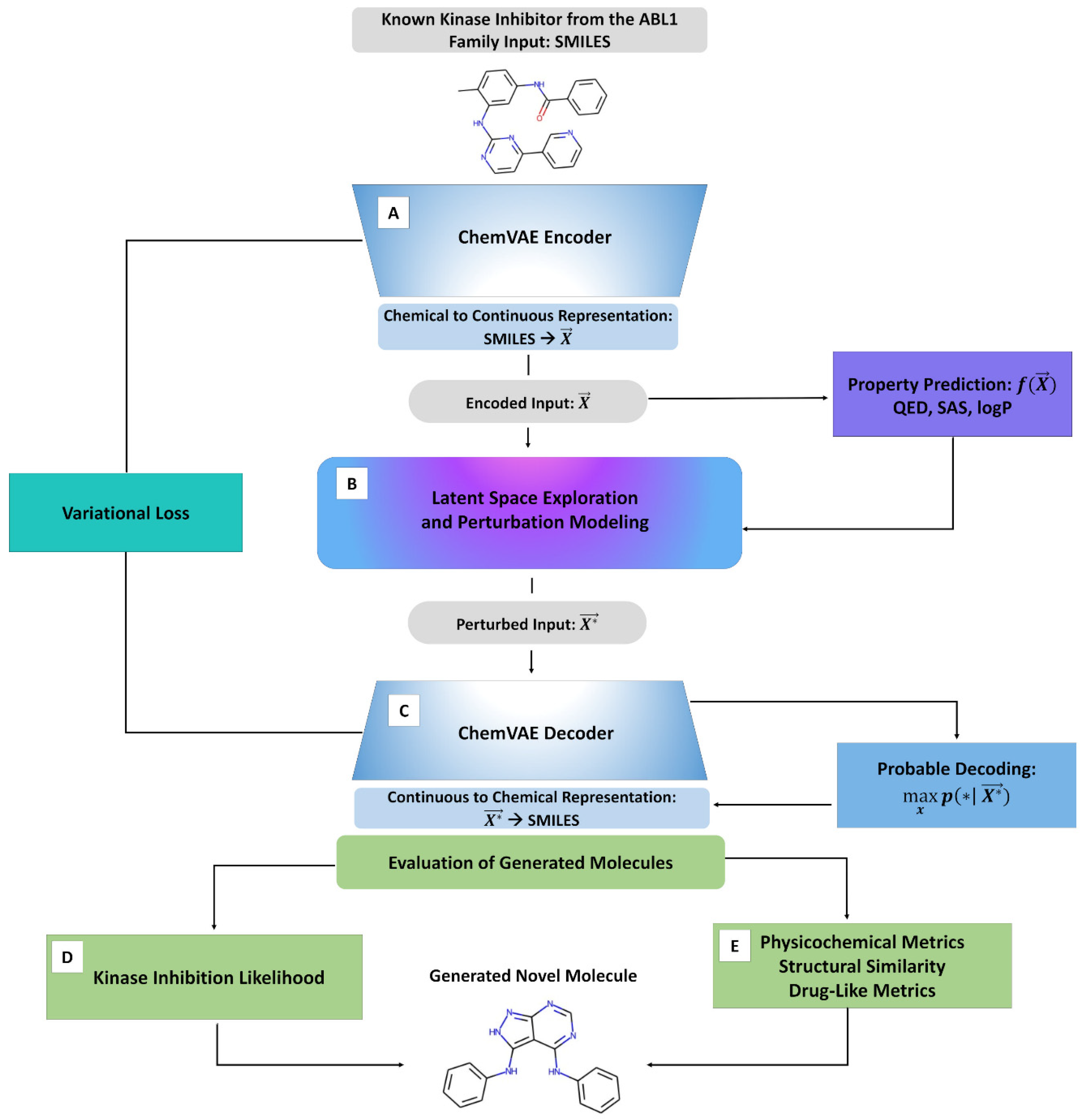
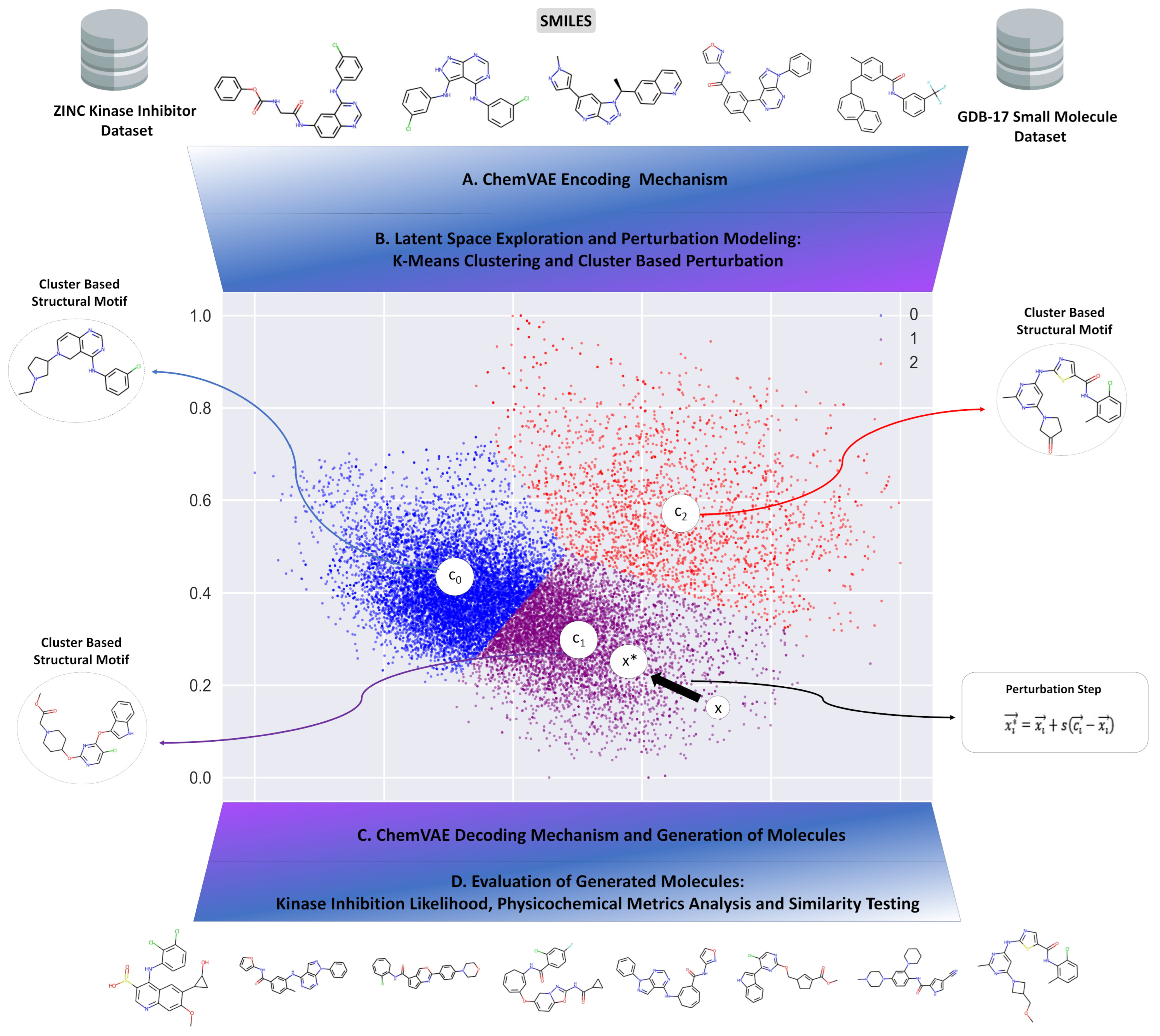
2.1. Integrative Machine Learning Model for Targeted Exploration of the Chemical Space
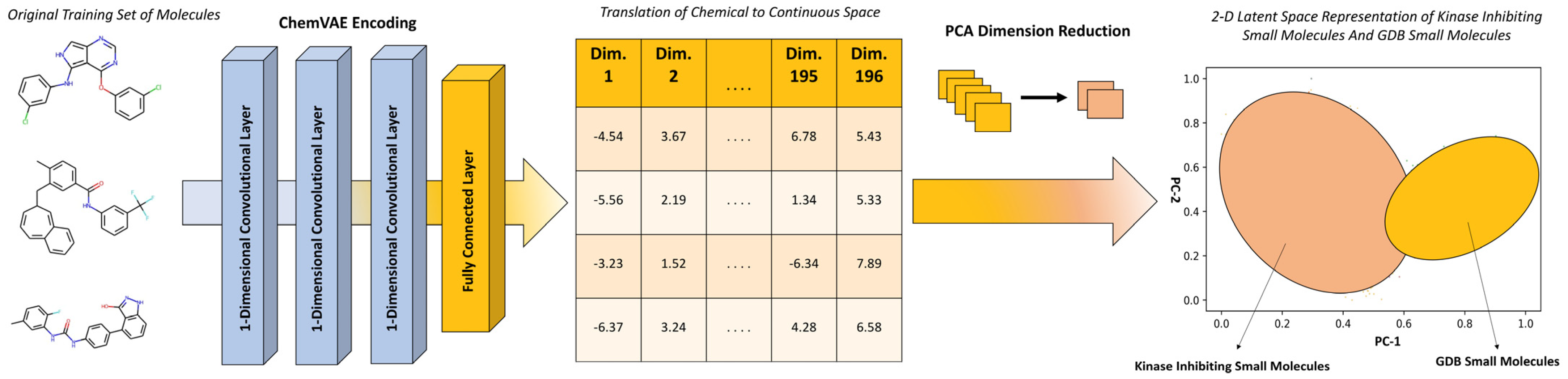
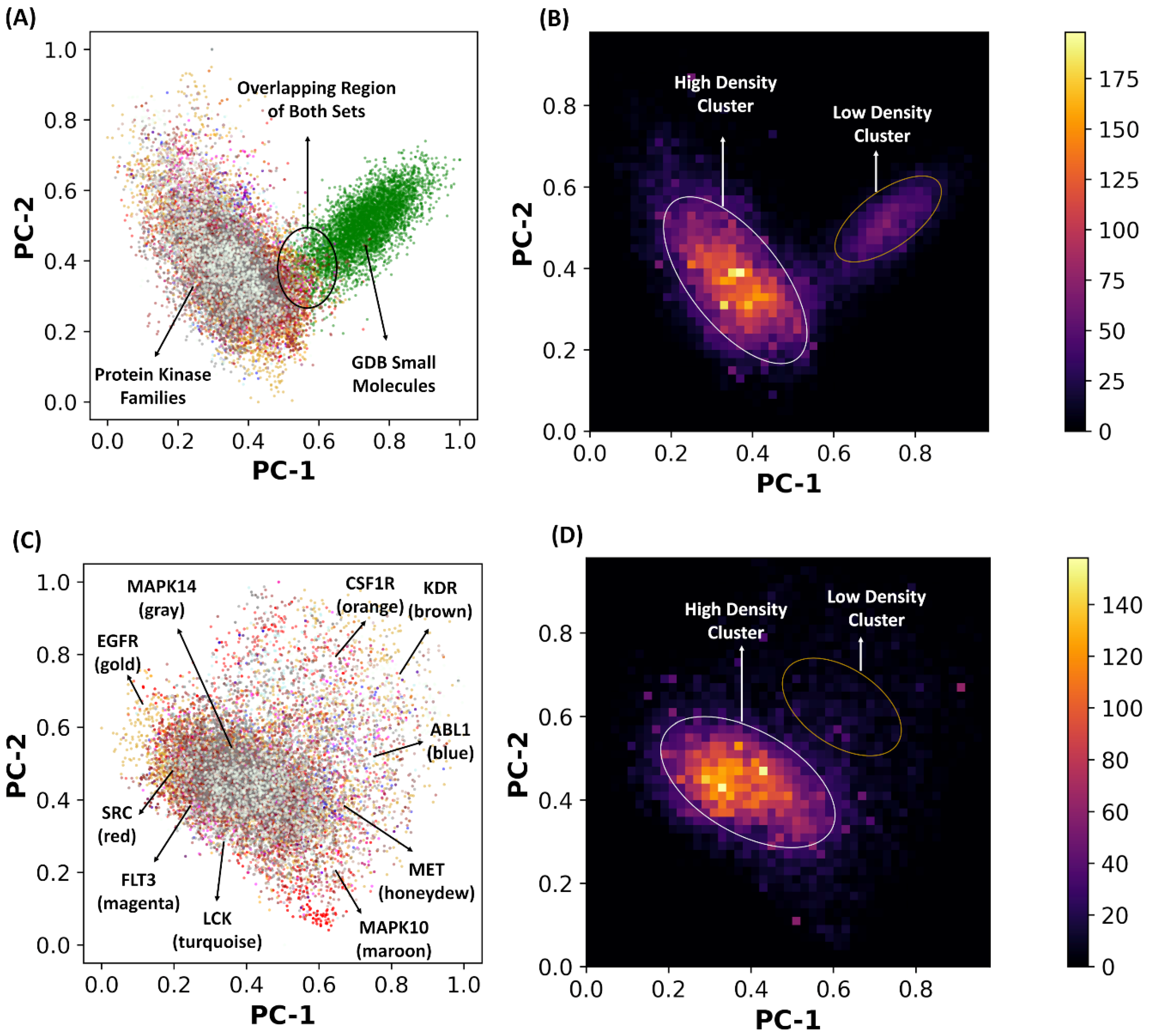
2.2. Principal Component Analysis of the Latent Space Landscapes for Small Molecules and Kinase Inhibitors
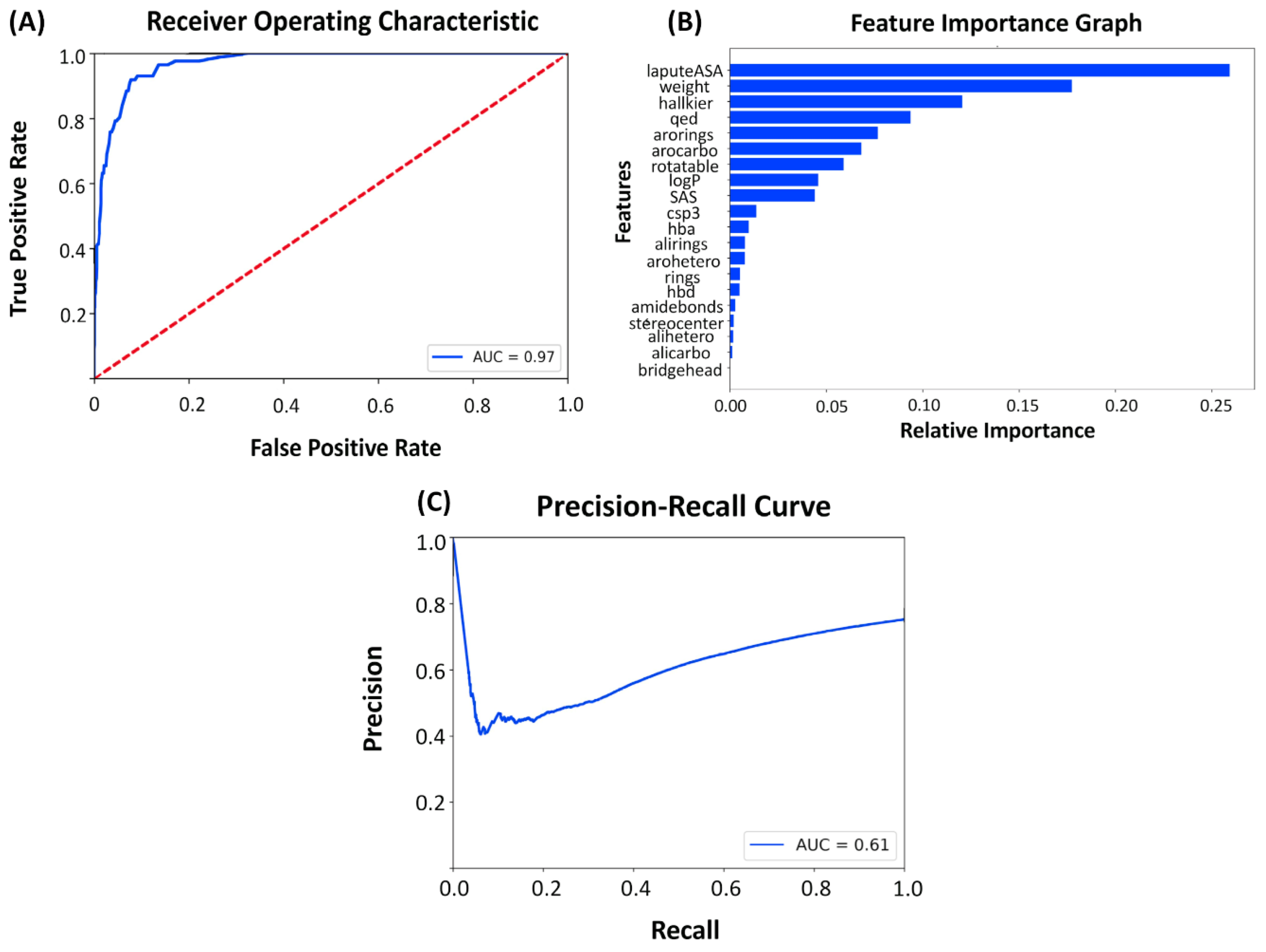
2.3. Latent Space and Chemical Feature-Based Kinase Inhibition Likelihood Classifiers
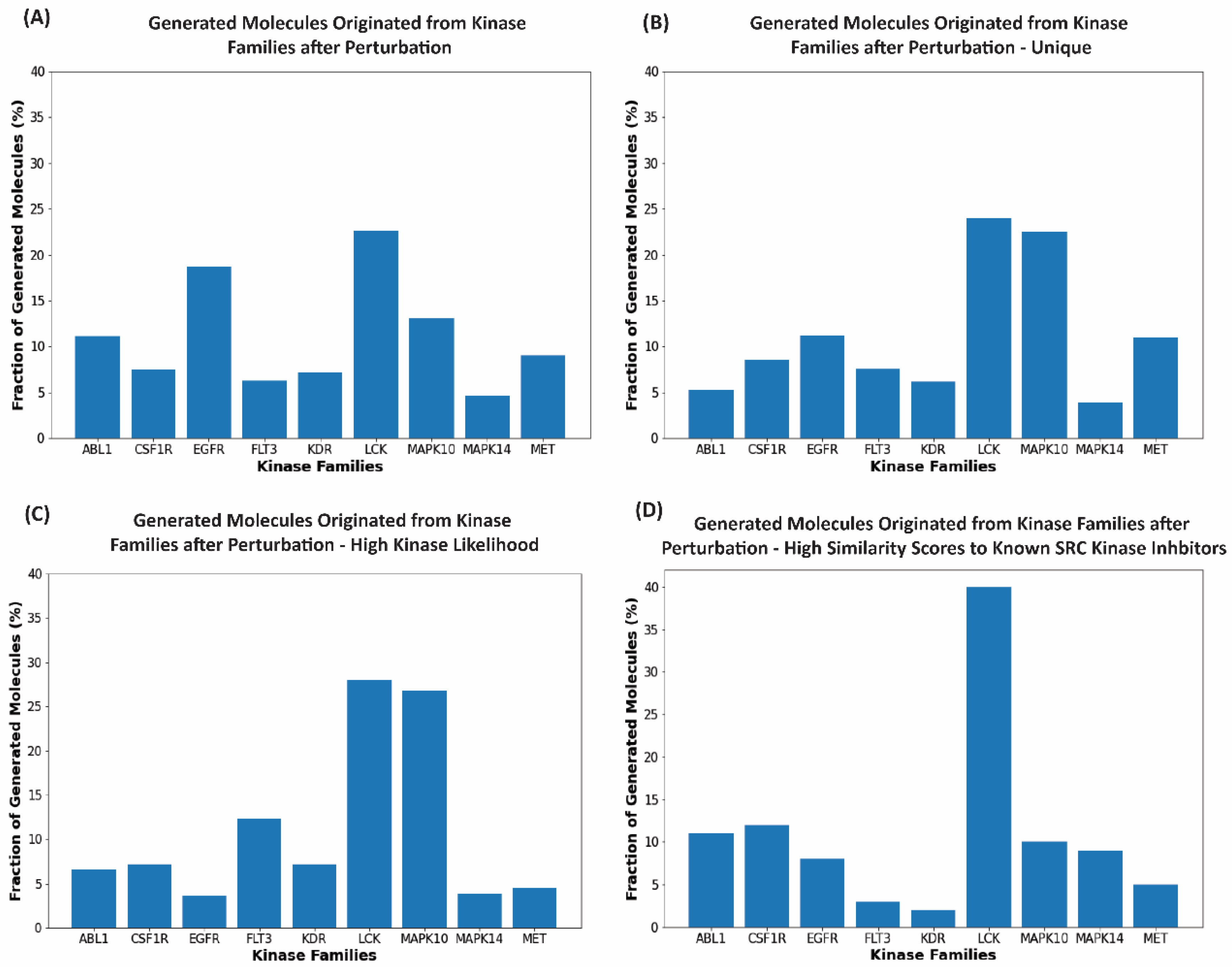
2.4. Cluster-Based Perturbation Approach for Targeted Exploration of the Latent Space and Molecular Design
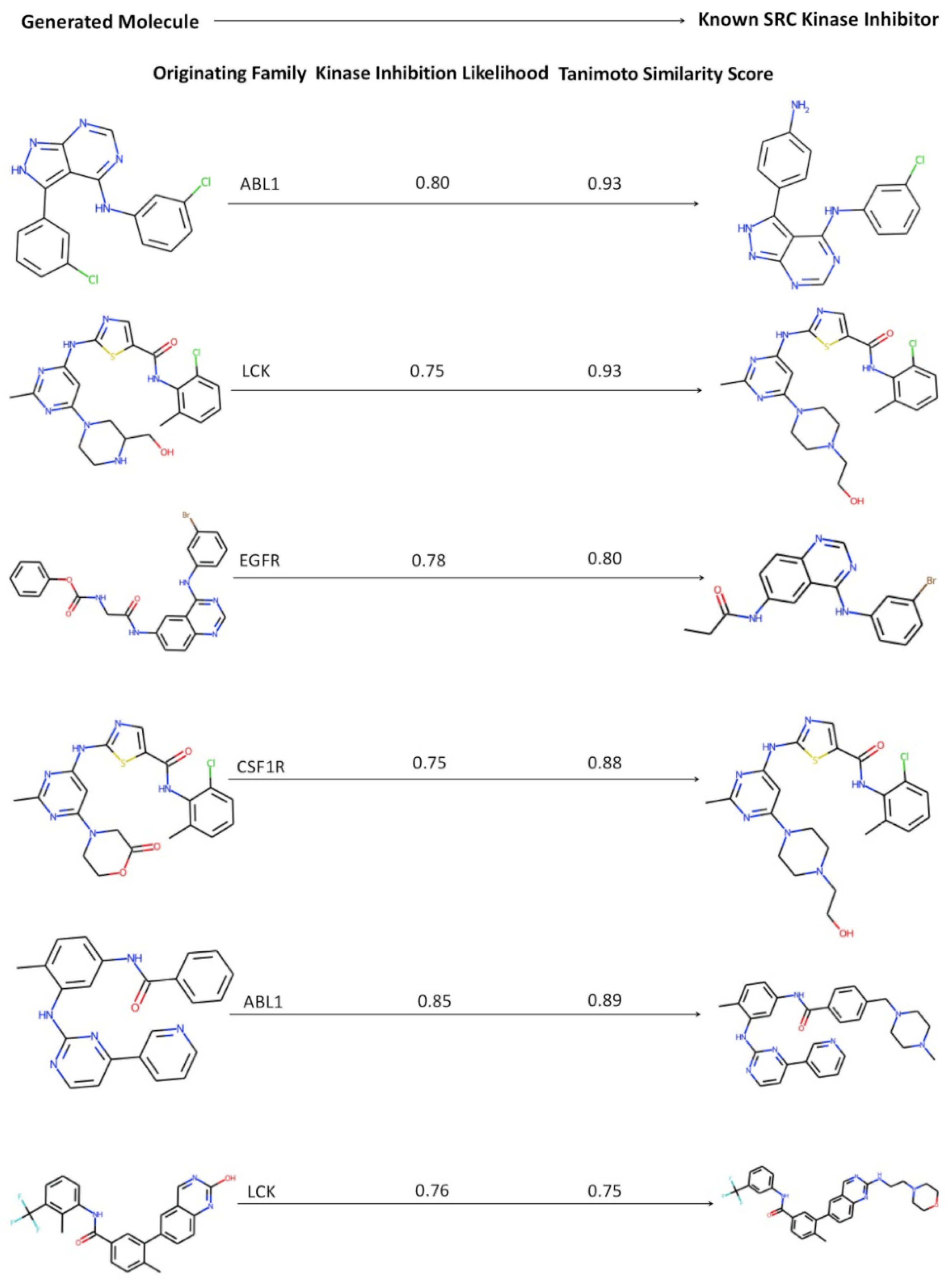
2.5. Kinase Inhibition Likelihood and Similarity Analyses of the Generated Molecules
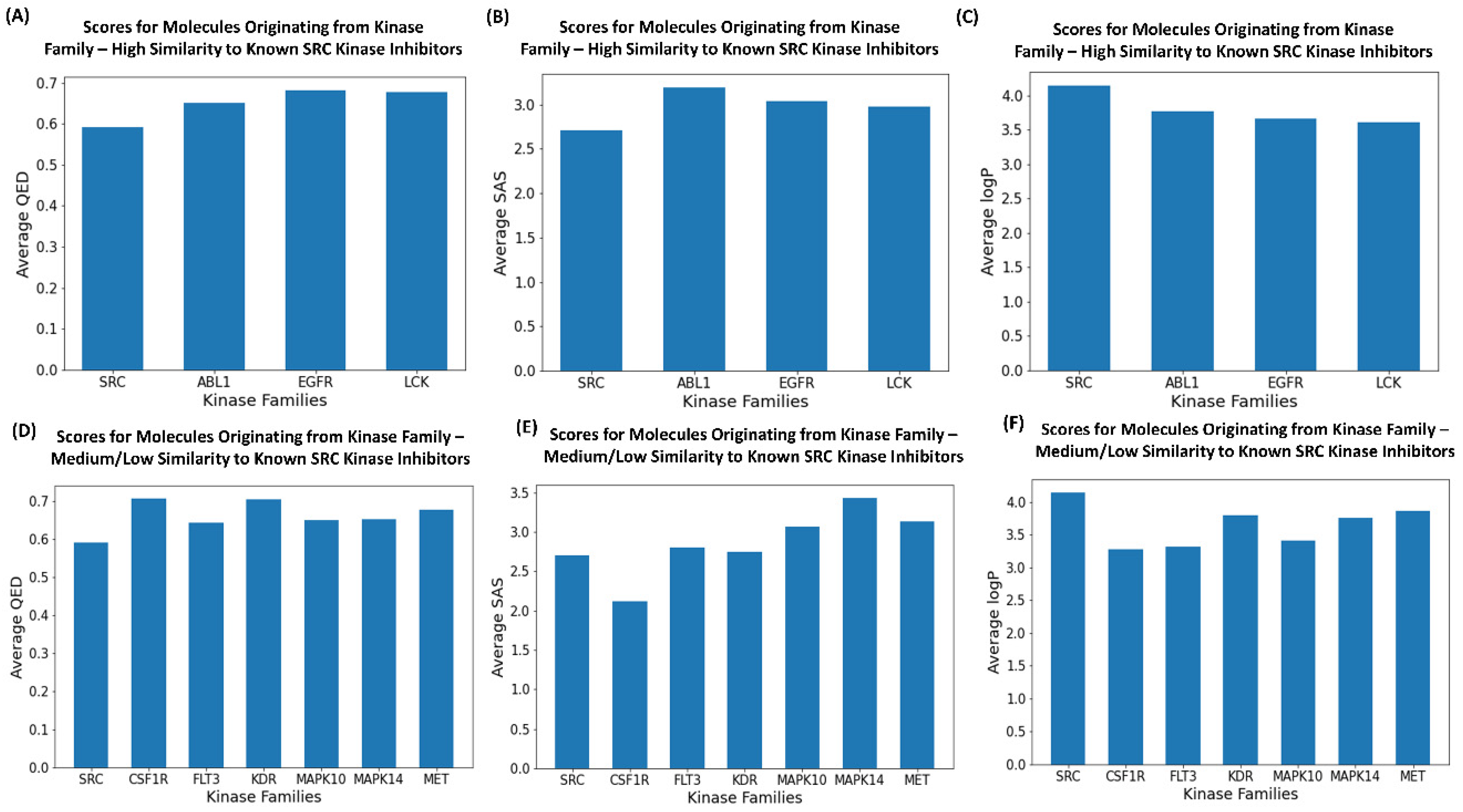
2.6. Drug-like Properties’ Assessment of the Generated Molecules
3. Materials and Methods
3.1. Datasets of Protein Kinase Inhibitors and Small Molecules
3.2. Clustering and Perturbation-Based Exploration of the Latent Space and Variational Autoencoder
3.3. Kinase Inhibition Likelihood Classifiers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mater, A.C.; Coote, M.L. Deep Learning in Chemistry. J. Chem. Inf. Model. 2019, 59, 2545–2559. [Google Scholar] [CrossRef]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep learning for computational chemistry. J. Comput Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, T.; Kreisbeck, C.; Becker, J.S.; Aspuru-Guzik, A.; Saikin, S.K. Autonomous Molecular Design: Then and Now. ACS Appl. Mater. Interfaces 2019, 11, 24825–24836. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Sousa, T.; Correia, J.; Pereira, V.; Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 2021, 61, 5343–5361. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P.; Bodor, N. Octanol-water partition: Searching for predictive models. Curr. Med. Chem. 1998, 5, 353–380. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. In Advances in Neural Information Processing Systems 27 (NIPS 2014); Curran Associates, Inc.: Red Hook, NY, USA, 2014; pp. 2672–2680. [Google Scholar]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced Adversarial Neural Computer for de Novo Molecular Design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar] [CrossRef]
- Maziarka, L.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Warchol, M. Mol-CycleGAN—A generative mode, for molecular optimization. J. Cheminform. 2020, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, K.; Wang, S.; Wang, L.; Cai, C.; Song, C.; Lai, L.; Pei, J. Deep learning for molecular generation. Future Med. Chem. 2019, 11, 567–597. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H. De Novo Molecule Design Using Molecular Generative Models Constrained by Ligand-Protein Interactions. J. Chem. Inf. Model. 2022, 62, 3291–3306. [Google Scholar] [CrossRef]
- Li, Y.; Pei, J.; Lai, L. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 2021, 12, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wang, F.; Li, Y.; Lai, L.; Pei, J. Advances and Challenges in De Novo Drug Design Using Three-Dimensional Deep Generative Models. J. Chem. Inf. Model. 2022, 62, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Dollar, O.; Joshi, N.; Beck, D.A.C.; Pfaendtner, J. Attention-based generative models for de novo molecular design. Chem. Sci. 2021, 12, 8362–8372. [Google Scholar] [CrossRef]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2018, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Montanari, F.; Steffen, A.; Briem, H.; Noé, F.; Clevert, D.A. Efficient multi-objective molecular optimization in a continuous latent space. Chem. Sci. 2019, 10, 8016–8024. [Google Scholar] [CrossRef]
- Winter, R.; Retel, J.; Noé, F.; Clevert, D.A.; Steffen, A. grünifai: Interactive multiparameter optimization of molecules in a continuous vector space. Bioinformatics 2020, 36, 4093–4094. [Google Scholar] [CrossRef]
- Hoffman, S.C.; Chenthamarakshan, V.; Wadhawan, K.; Cen, P.-Y.; Das, P. Optimizing molecules using efficient queries from property evaluations. Nat. Mach. Intell. 2022, 4, 21–31. [Google Scholar] [CrossRef]
- Wang, M.; Sun, H.; Wang, J.; Pang, J.; Chai, X.; Xu, L.; Li, H.; Cao, D.; Hou, T. Comprehensive assessment of deep generative architectures for de novo drug design. Brief. Bioinform. 2022, 23, bbab544. [Google Scholar] [CrossRef]
- Yang, M.; Tao, B.; Chen, C.; Jia, W.; Sun, S.; Zhang, T.; Wang, X. Machine Learning Models Based on Molecular Fingerprints and an Extreme Gradient Boosting Method Lead to the Discovery of JAK2 Inhibitors. J. Chem. Inf. Model. 2019, 59, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, R.; Bajorath, J. Multitask machine learning for classifying highly and weakly potent kinase inhibitors. ACS Omega 2019, 4, 4367–4375. [Google Scholar] [CrossRef]
- Zhang, Z.; Guan, J.; Zhou, S. FraGAT: A fragment-oriented multi-scale graph attention model for molecular property prediction. Bioinformatics 2021, 37, 2981–2987. [Google Scholar] [CrossRef]
- Jiang, D.; Wu, Z.; Hsieh, C.Y.; Chen, G.; Liao, B.; Wang, Z.; Shen, C.; Cao, D.; Wu, J.; Hou, T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 2021, 13, 12. [Google Scholar] [CrossRef]
- Shen, W.X.; Zeng, X.; Zhu, F.; Wang, Y.L.; Qin, C.; Tan, Y.; Jiang, Y.Y.; Chen, Y.Z. Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations. Nat. Mach. Intell. 2021, 3, 334–343. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Yoshimori, A.; Miljković, F.; Bajorath, J. Approach for the Design of Covalent Protein Kinase Inhibitors via Focused Deep Generative Modeling. Molecules 2022, 27, 570. [Google Scholar] [CrossRef]
- Feldmann, C.; Bajorath, J. Differentiating Inhibitors of Closely Related Protein Kinases with Single- or Multi-Target Activity via Explainable Machine Learning and Feature Analysis. Biomolecules 2022, 12, 557. [Google Scholar] [CrossRef]
- Miljković, F.; Rodríguez-Pérez, R.; Bajorath, J. Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J. Med. Chem. 2020, 63, 8738–8748. [Google Scholar] [CrossRef]
- Abdelbaky, I.; Tayara, H.; Chong, K.T. Prediction of kinase inhibitors binding modes with machine learning and reduced descriptor sets. Sci Rep. 2021, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B., Jr.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein-ligand binding affinities and structures. Nucleic Acids Res. 2015, 43, D465–D469. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; de Matos, P.; Dekker, A.; Ennis, M.; Harsha, B.; Kale, N.; Muthukrishnan, V.; Owen, G.; Turner, S.; Williams, M.; et al. The ChEBI reference database and ontology for biologically relevant chemistry: Enhancements for 2013. Nucleic Acids Res. 2013, 41, D456–D463. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Hu, H.; Laufkötter, O.; Miljković, F.; Bajorath, J. Data set of competitive and allosteric protein kinase inhibitors confirmed by X-ray crystallography. Data Brief. 2021, 35, 106816. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Visini, R.; Awale, M.; Reymond, J.L. Fragment Database FDB-17. J. Chem. Inf. Model. 2017, 57, 700–709. [Google Scholar] [CrossRef]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An open source chemical structure curation pipeline using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Godden, J.W.; Xue, L.; Bajorath, J. Combinatorial preferences affect molecular similarity/diversity calculations using binary fingerprints and Tanimoto coefficients. J. Chem. Inf. Comput. Sci. 2000, 40, 163–166. [Google Scholar] [CrossRef]
- Bournez, C.; Carles, F.; Peyrat, G.; Aci-Sèche, S.; Bourg, S.; Meyer, C.; Bonnet, P. Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB. Molecules 2020, 25, 3226. [Google Scholar] [CrossRef] [PubMed]
- Likas, A.; Vlassis, N.; Verbeek, J.J. The global k-means clustering algorithm. Pattern Recognit. 2003, 36, 451–461. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; Volume 16, pp. 265–283. [CrossRef]
- Boulesteix, A.; Janitza, S.; Kruppa, J.; König, I. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Data Min. Knowl. Discov. 2012, 2, 493–507. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug-Like Metrics | SRC Inhibitors | ABL1 Inhibitors | LCK Inhibitors | EGFR Inhibitors |
|---|---|---|---|---|
| Average QED Scores | 0.605 | 0.6742 | 0.6952 | 0.6752 |
| Average SAS Score | 2.6829 | 2.9806 | 2.012 | 2.8175 |
| Average logP Scores | 3.869 | 3.8989 | 3.803 | 3.7687 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, K.; Kassab, R.; Agajanian, S.; Verkhivker, G. Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration. Int. J. Mol. Sci. 2022, 23, 11262. https://doi.org/10.3390/ijms231911262
Krishnan K, Kassab R, Agajanian S, Verkhivker G. Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration. International Journal of Molecular Sciences. 2022; 23(19):11262. https://doi.org/10.3390/ijms231911262
Chicago/Turabian StyleKrishnan, Keerthi, Ryan Kassab, Steve Agajanian, and Gennady Verkhivker. 2022. "Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration" International Journal of Molecular Sciences 23, no. 19: 11262. https://doi.org/10.3390/ijms231911262
APA StyleKrishnan, K., Kassab, R., Agajanian, S., & Verkhivker, G. (2022). Interpretable Machine Learning Models for Molecular Design of Tyrosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration. International Journal of Molecular Sciences, 23(19), 11262. https://doi.org/10.3390/ijms231911262