
The Structural Effects of Phosphorylation of Protein Arginine Methyltransferase 5 on Its Binding to Histone H4

 and
and 
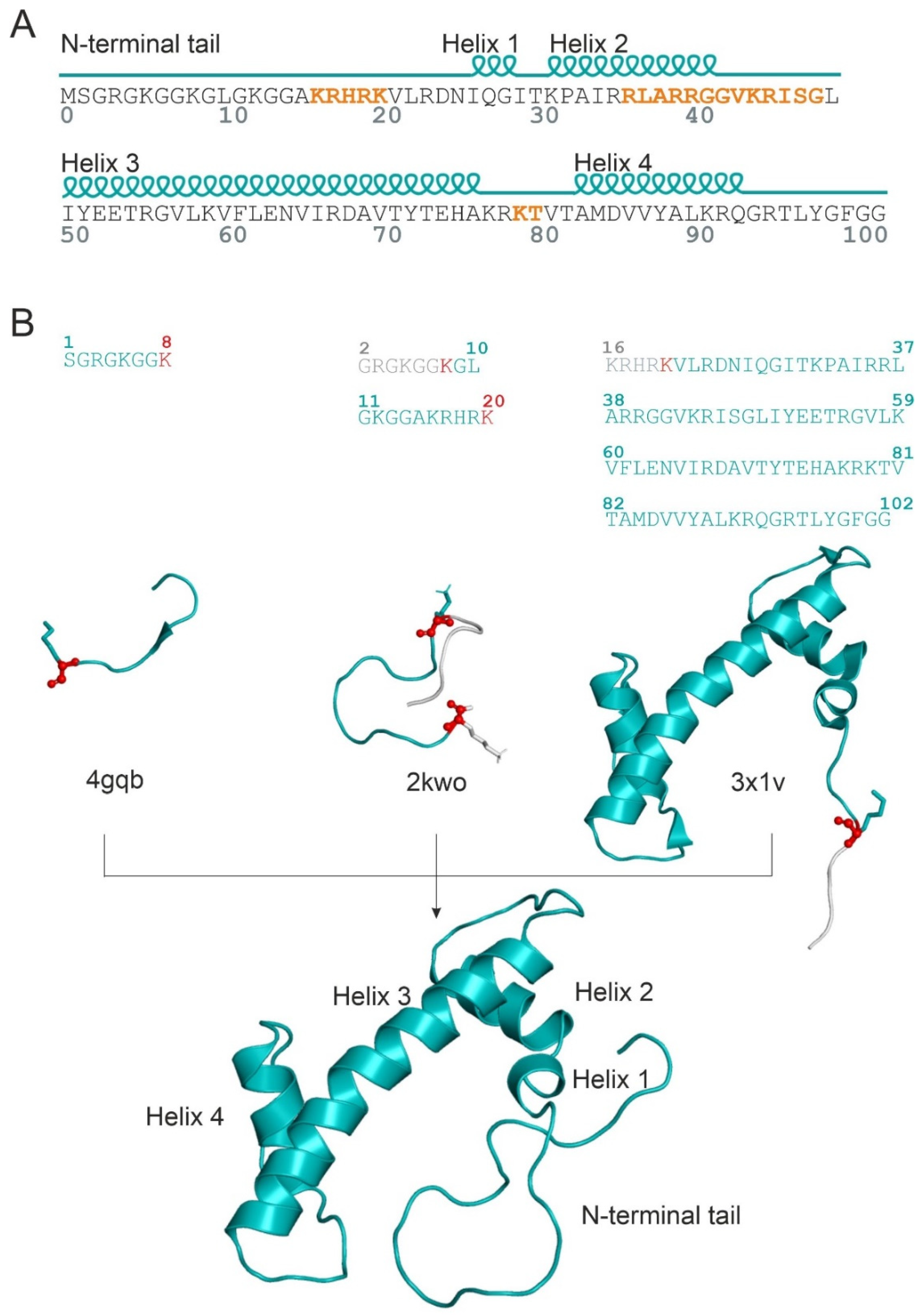{kind=link}
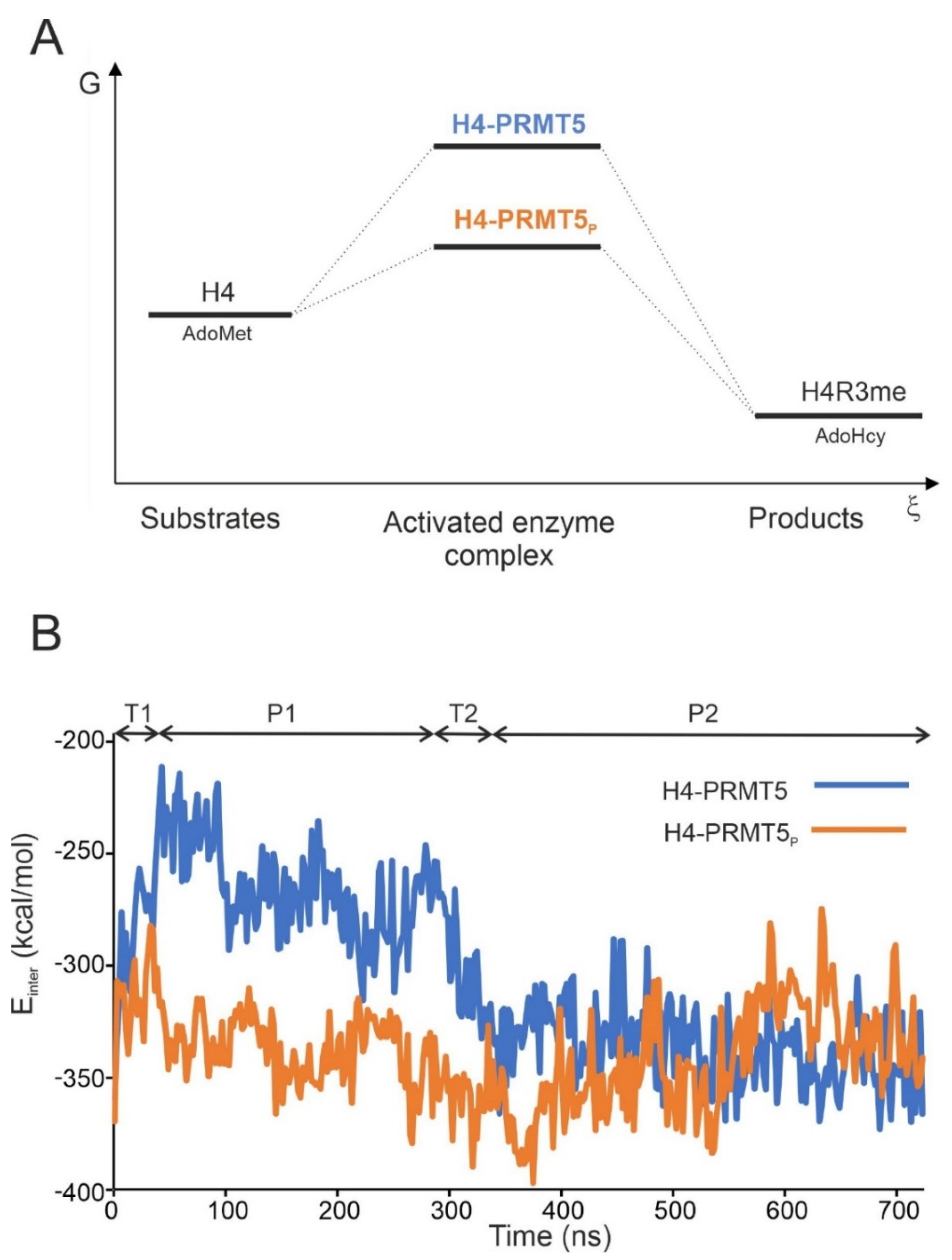{kind=link}
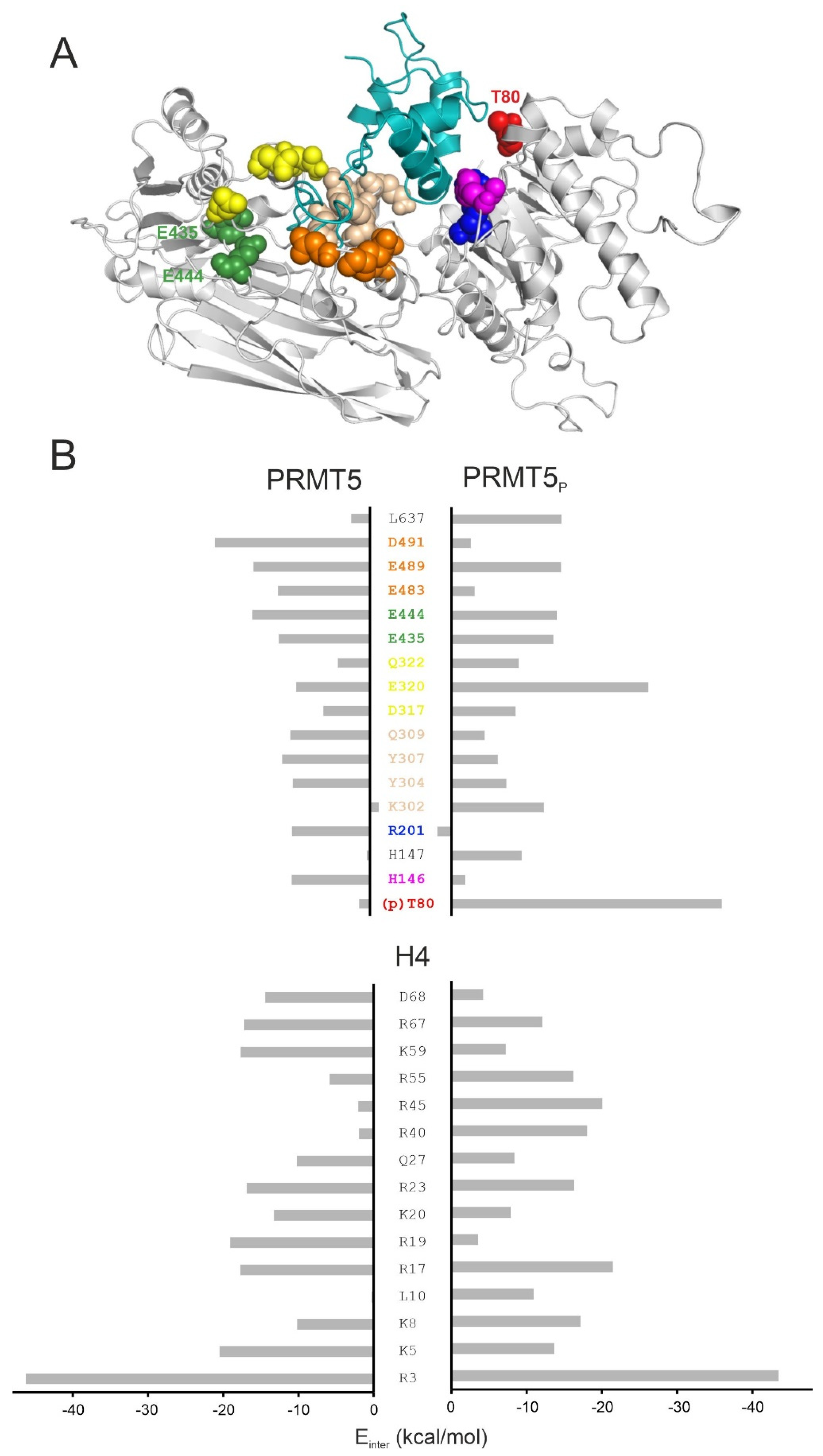{kind=link}
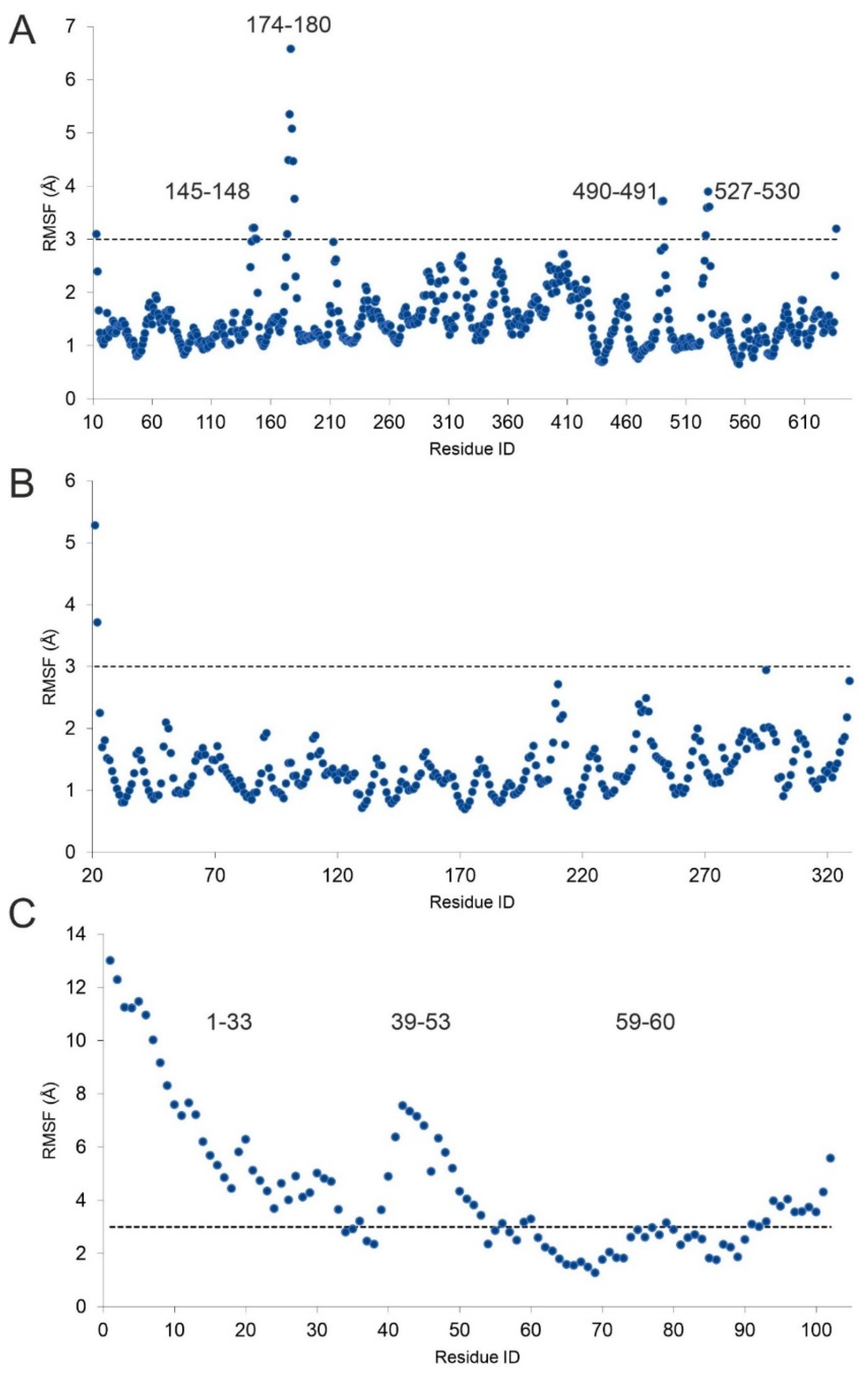{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Unmodified PRMT5 in Complex with the Full-Length H4 Protein
2.2. The H4-PRMT5-MEP Complex vs. Apo Protein Structures
2.3. Structural Effects of Phosphorylation on T80
3. Materials and Methods
3.1. In Situ Fragment-Based Construction of H4 in Complex with PRMT5
3.2. Energy Minimization
3.3. Molecular Dynamic Simulation
3.4. Interaction Energy Calculations
3.5. Selection of Representative Structures by Structural Clustering and Interaction Energy Differences
3.6. Determination of DNA Binding Domain of H4 in Nucleosome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PTM | Post-translational modification |
| PRMT | Protein arginine methyltransferase |
| PRMT5 | Protein arginine methyltransferase 5 |
| H4 | Histone H4 peptide |
| PRMT5P | Human PRMT5 enzyme phosphorylated on T80 residue |
| pT80 | Phosphorylated T80 residue |
| MEP50 | Methylosome protein 50 |
| G | Free energy |
| Einter | The sum of Coulomb and Lennard-Jones intermolecular interaction energy |
| MD | Molecular dynamics |
| PDB | Protein Data Bank |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
References
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Izzo, L.T.; Wellen, K.E. Histone lactylation links metabolism and gene regulation. Nature 2019, 574, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; Dann, G.P.; Beh, L.Y.; Debelouchina, G.T.; Hofmann, R.; Muir, T.W. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun. 2018, 9, 1394. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Hetényi, C. Molecular Structure, Binding Affinity, and Biological Activity in the Epigenome. Int. J. Mol. Sci. 2020, 21, 4134. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Deindl, S.; Harada, B.T.; Zhuang, X. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature 2014, 512, 213–217. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Allison, K.A.; Kaikkonen, M.U.; Orozco, L.D.; Glass, C.K. Impact of natural genetic variation on enhancer selection and function. Nature 2013, 503, 487–492. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Sipos, A.; Iván, J.; Bécsi, B.; Darula, Z.; Tamás, I.; Horváth, D.; Medzihradszky, K.F.; Erdődi, F.; Lontay, B. Myosin phosphatase and RhoA-activated kinase modulate arginine methylation by the regulation of protein arginine methyltransferase 5 in hepatocellular carcinoma cells. Sci. Rep. 2017, 7, 40590. [Google Scholar] [CrossRef]
- Ancelin, K.; Lange, U.C.; Hajkova, P.; Schneider, R.; Bannister, A.J.; Kouzarides, T.; Surani, M.A. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat. Cell Biol. 2006, 8, 623–630. [Google Scholar] [CrossRef]
- Fulton, M.D.; Cao, M.; Ho, M.-C.; Zhao, X.; Zheng, Y.G. The macromolecular complexes of histones affect protein arginine methyltransferase activities. J. Biol. Chem. 2021, 297, 101123. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Li, Q.; Yang, C.; Huo, D.; Wang, X.; Ai, C.; Kong, Y.; Sun, X.; Wang, W.; Zhou, Y.; et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat. Commun. 2018, 9, 4552. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef]
- Antonysamy, S. The Structure and Function of the PRMT5:MEP50 Complex. In Macromolecular Protein Complexes; Harris, J.R., Marles-Wright, J., Eds.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2017; Volume 83, pp. 185–194. ISBN 978-3-319-46501-2. [Google Scholar]
- Burgos, E.S.; Wilczek, C.; Onikubo, T.; Bonanno, J.B.; Jansong, J.; Reimer, U.; Shechter, D. Histone H2A and H4 N-terminal Tails Are Positioned by the MEP50 WD Repeat Protein for Efficient Methylation by the PRMT5 Arginine Methyltransferase *. J. Biol. Chem. 2015, 290, 9674–9689. [Google Scholar] [CrossRef]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-mediated phosphorylation of PRMT5 down-regulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef]
- Lattouf, H.; Kassem, L.; Jacquemetton, J.; Choucair, A.; Poulard, C.; Trédan, O.; Corbo, L.; Diab-Assaf, M.; Hussein, N.; Treilleux, I.; et al. LKB1 regulates PRMT5 activity in breast cancer. Int. J. Cancer 2019, 144, 595–606. [Google Scholar] [CrossRef]
- Ho, M.-C.; Wilczek, C.; Bonanno, J.B.; Xing, L.; Seznec, J.; Matsui, T.; Carter, L.G.; Onikubo, T.; Kumar, P.R.; Chan, M.K.; et al. Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS ONE 2013, 8, e57008. [Google Scholar] [CrossRef]
- Sun, L.; Wang, M.; Lv, Z.; Yang, N.; Liu, Y.; Bao, S.; Gong, W.; Xu, R.-M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. USA 2011, 108, 20538–20543. [Google Scholar] [CrossRef]
- Kawamura, S.; Palte, R.L.; Kim, H.-Y.; Saurí, J.; Sondey, C.; Mansueto, M.S.; Altman, M.D.; Machacek, M.R. Design and Synthesis of Unprecedented 9- and 10-Membered Cyclonucleosides with PRMT5 Inhibitory Activity. Bioorg. Med. Chem. 2022, 66, 116820. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Aranda, R.; Bobinski, T.P.; Briere, D.M.; Burns, A.C.; Christensen, J.G.; Clarine, J.; Engstrom, L.D.; Gunn, R.J.; Ivetac, A.; et al. Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5 MTA Complex for the Treatment of MTAP-Deleted Cancers. J. Med. Chem. 2022, 65, 1749–1766. [Google Scholar] [CrossRef]
- Jensen-Pergakes, K.; Tatlock, J.; Maegley, K.A.; McAlpine, I.J.; McTigue, M.; Xie, T.; Dillon, C.P.; Wang, Y.; Yamazaki, S.; Spiegel, N.; et al. SAM-Competitive PRMT5 Inhibitor PF-06939999 Demonstrates Antitumor Activity in Splicing Dysregulated NSCLC with Decreased Liability of Drug Resistance. Mol. Cancer Ther. 2022, 21, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, R.V.; Reutershan, M.H.; Schneider, S.E.; Sloman, D.; Lacey, B.M.; Swalm, B.M.; Yeung, C.S.; Gibeau, C.; Spellman, D.S.; Rankic, D.A.; et al. The Discovery of Two Novel Classes of 5,5-Bicyclic Nucleoside-Derived PRMT5 Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 3911–3939. [Google Scholar] [CrossRef]
- Candito, D.A.; Ye, Y.; Quiroz, R.V.; Reutershan, M.H.; Witter, D.; Gadamsetty, S.B.; Li, H.; Saurí, J.; Schneider, S.E.; Lam, Y.; et al. Development of a Flexible and Robust Synthesis of Tetrahydrofuro[3,4-b]Furan Nucleoside Analogues. J. Org. Chem. 2021, 86, 5142–5151. [Google Scholar] [CrossRef]
- McKinney, D.C.; McMillan, B.J.; Ranaghan, M.J.; Moroco, J.A.; Brousseau, M.; Mullin-Bernstein, Z.; O’Keefe, M.; McCarren, P.; Mesleh, M.F.; Mulvaney, K.M.; et al. Discovery of a First-in-Class Inhibitor of the PRMT5–Substrate Adaptor Interaction. J. Med. Chem. 2021, 64, 11148–11168. [Google Scholar] [CrossRef]
- Mulvaney, K.M.; Blomquist, C.; Acharya, N.; Li, R.; Ranaghan, M.J.; O’Keefe, M.; Rodriguez, D.J.; Young, M.J.; Kesar, D.; Pal, D.; et al. Molecular Basis for Substrate Recruitment to the PRMT5 Methylosome. Mol. Cell 2021, 81, 3481–2495.e7. [Google Scholar] [CrossRef]
- Palte, R.L.; Schneider, S.E.; Altman, M.D.; Hayes, R.P.; Kawamura, S.; Lacey, B.M.; Mansueto, M.S.; Reutershan, M.; Siliphaivanh, P.; Sondey, C.; et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med. Chem. Lett. 2020, 11, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, M.; Zhang, Y.W.; Tong, S.; Leal, R.A.; Shetty, R.; Vaddi, K.; Luengo, J.I. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033–1038. [Google Scholar] [CrossRef]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; McDonald, E.R.; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered Methionine Metabolism in MTAP/CDKN2A-Deleted Cancers Leads to Dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A Selective Inhibitor of PRMT5 with in Vivo and in Vitro Potency in MCL Models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9Å Resolution††We dedicate this paper to the memory of Max Perutz who was particularly inspirational and supportive to T.J.R. in the early stages of this study. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Abdelsattar, A.S.; Mansour, Y.; Aboul-ela, F. The Perturbed Free-Energy Landscape: Linking Ligand Binding to Biomolecular Folding. ChemBioChem 2021, 22, 1499–1516. [Google Scholar] [CrossRef]
- Fu, I.; Geacintov, N.E.; Broyde, S. Molecular dynamics simulations reveal how H3K56 acetylation impacts nucleosome structure to promote DNA exposure for lesion sensing. DNA Repair 2021, 107, 103201. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Laskowski, R.A.; Thornton, J.M. Amino acid–base interactions: A three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical Biology of Protein Arginine Modifications in Epigenetic Regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B. The UniProt Consortium UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Li, S.; Plotnikov, A.N.; Walsh, M.J.; Zhou, M.-M. Mechanism and Regulation of Acetylated Histone Binding by the Tandem PHD Finger of DPF3b. Nature 2010, 466, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Padavattan, S.; Shinagawa, T.; Hasegawa, K.; Kumasaka, T.; Ishii, S.; Kumarevel, T. Structural and functional analyses of nucleosome complexes with mouse histone variants TH2a and TH2b, involved in reprogramming. Biochem. Biophys. Res. Commun. 2015, 464, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017–4: Maestro; Schrödinger, LLC: New York, NY, USA, 2017.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER force-field parameters for phosphorylated amino acids in different protonation states: Phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Horváth, I.; Jeszenői, N.; Bálint, M.; Paragi, G.; Hetényi, C. A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level. Int. J. Mol. Sci. 2019, 20, E4384. [Google Scholar] [CrossRef]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. Des. Sel. 1991, 4, 903–910. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Li, J.; Cai, Q.; Hsieh, M.; Luo, R.; Duan, Y. Development of Polarizable Models for Molecular Mechanical Calculations IV: Van der Waals parameterization. J. Phys. Chem. B 2012, 116, 7088–7101. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Börzsei, R.; Bayarsaikhan, B.; Zsidó, B.Z.; Lontay, B.; Hetényi, C. The Structural Effects of Phosphorylation of Protein Arginine Methyltransferase 5 on Its Binding to Histone H4. Int. J. Mol. Sci. 2022, 23, 11316. https://doi.org/10.3390/ijms231911316
Börzsei R, Bayarsaikhan B, Zsidó BZ, Lontay B, Hetényi C. The Structural Effects of Phosphorylation of Protein Arginine Methyltransferase 5 on Its Binding to Histone H4. International Journal of Molecular Sciences. 2022; 23(19):11316. https://doi.org/10.3390/ijms231911316
Chicago/Turabian StyleBörzsei, Rita, Bayartsetseg Bayarsaikhan, Balázs Zoltán Zsidó, Beáta Lontay, and Csaba Hetényi. 2022. "The Structural Effects of Phosphorylation of Protein Arginine Methyltransferase 5 on Its Binding to Histone H4" International Journal of Molecular Sciences 23, no. 19: 11316. https://doi.org/10.3390/ijms231911316
APA StyleBörzsei, R., Bayarsaikhan, B., Zsidó, B. Z., Lontay, B., & Hetényi, C. (2022). The Structural Effects of Phosphorylation of Protein Arginine Methyltransferase 5 on Its Binding to Histone H4. International Journal of Molecular Sciences, 23(19), 11316. https://doi.org/10.3390/ijms231911316