

Decreased Expression of the Slc31a1 Gene and Cytoplasmic Relocalization of Membrane CTR1 Protein in Renal Epithelial Cells: A Potent Protective Mechanism against Copper Nephrotoxicity in a Mouse Model of Menkes Disease
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Copper Concentration in the Liver, Kidney and Urine of Experimental Mice
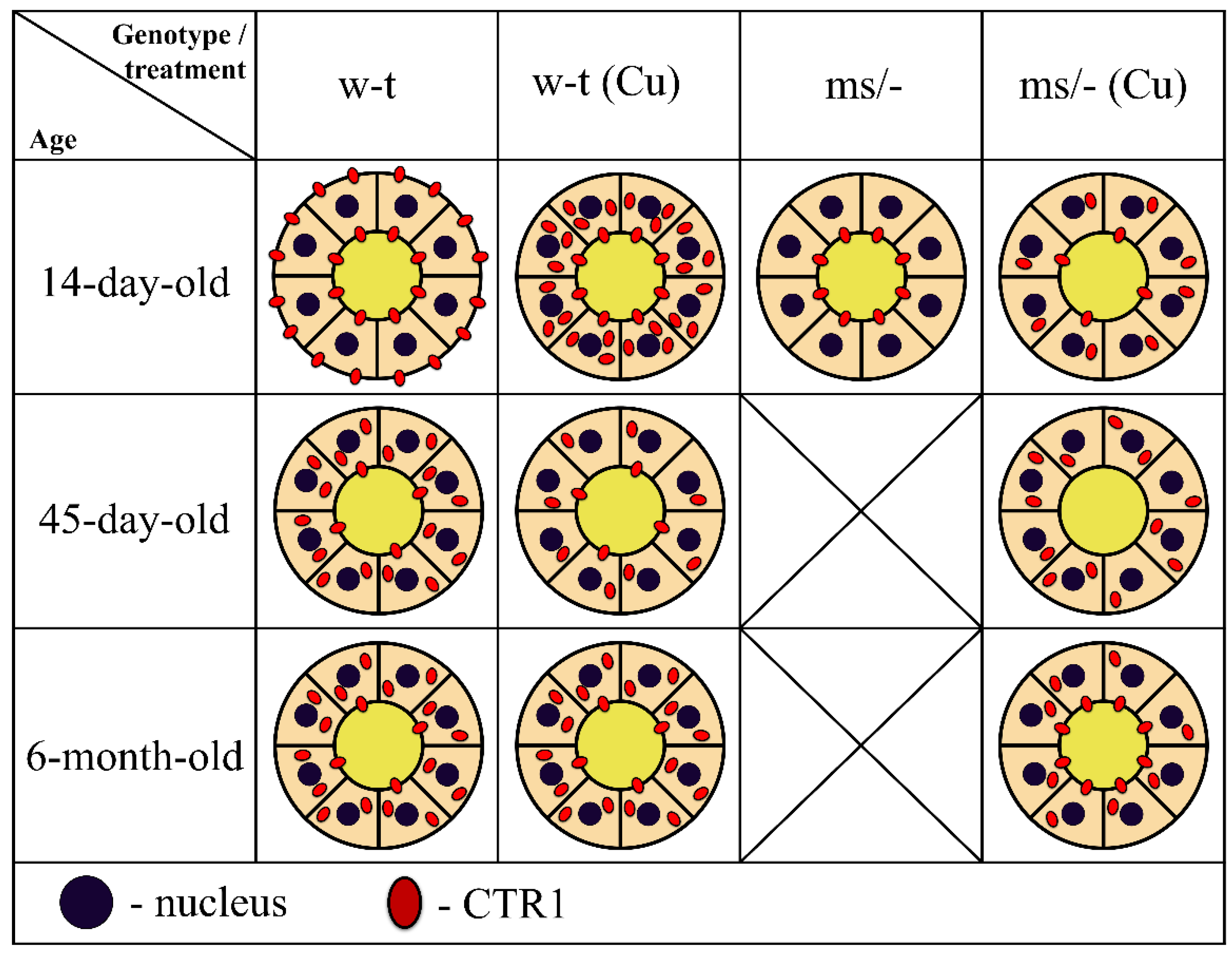
2.2. Effect of Copper Supplementation on the Renal Expression of Slc31a1 and Slc3121 Genes at the mRNA and Protein Levels in 14-Day-Old Mosaic Mutant and W-T Mice
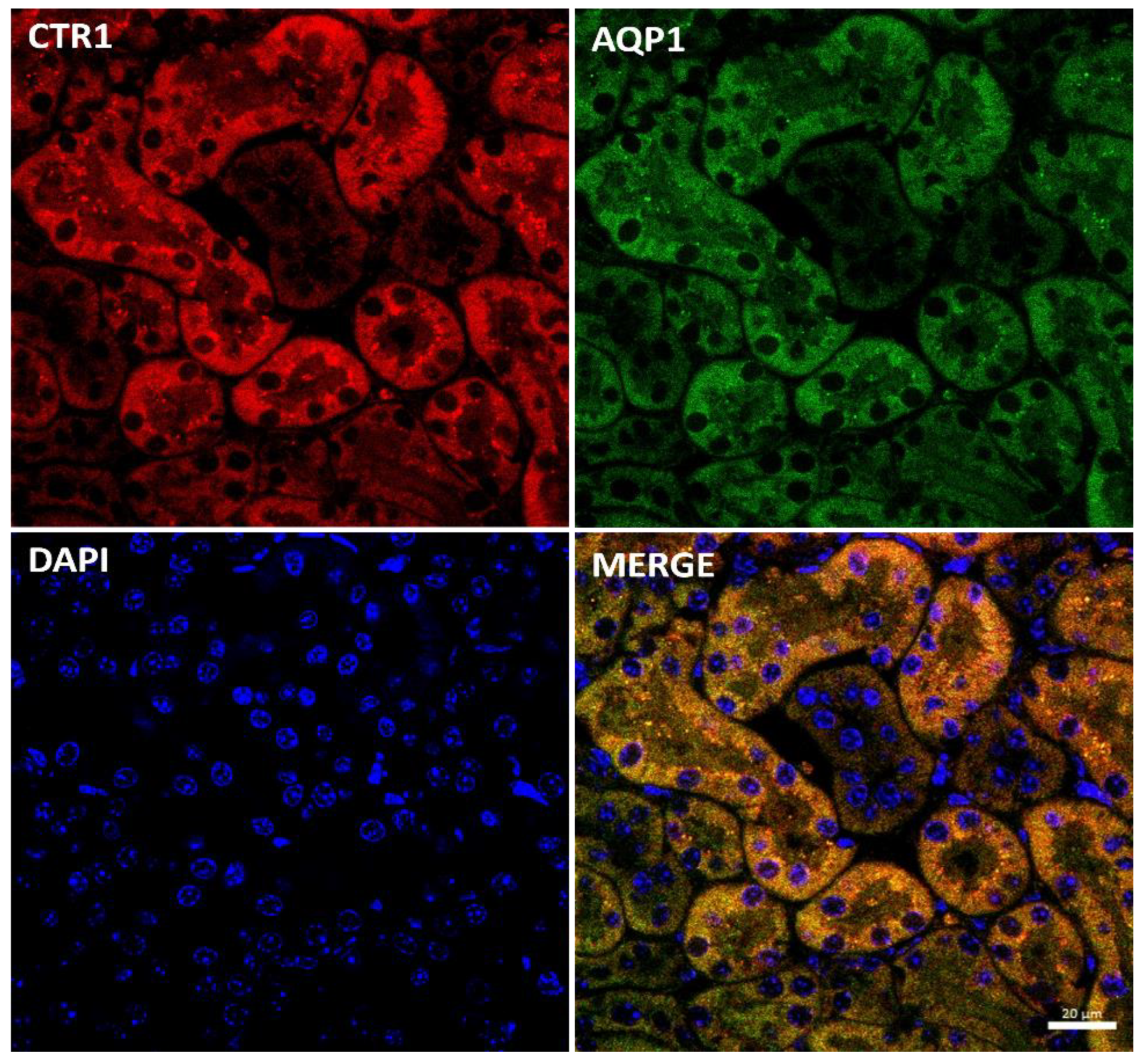
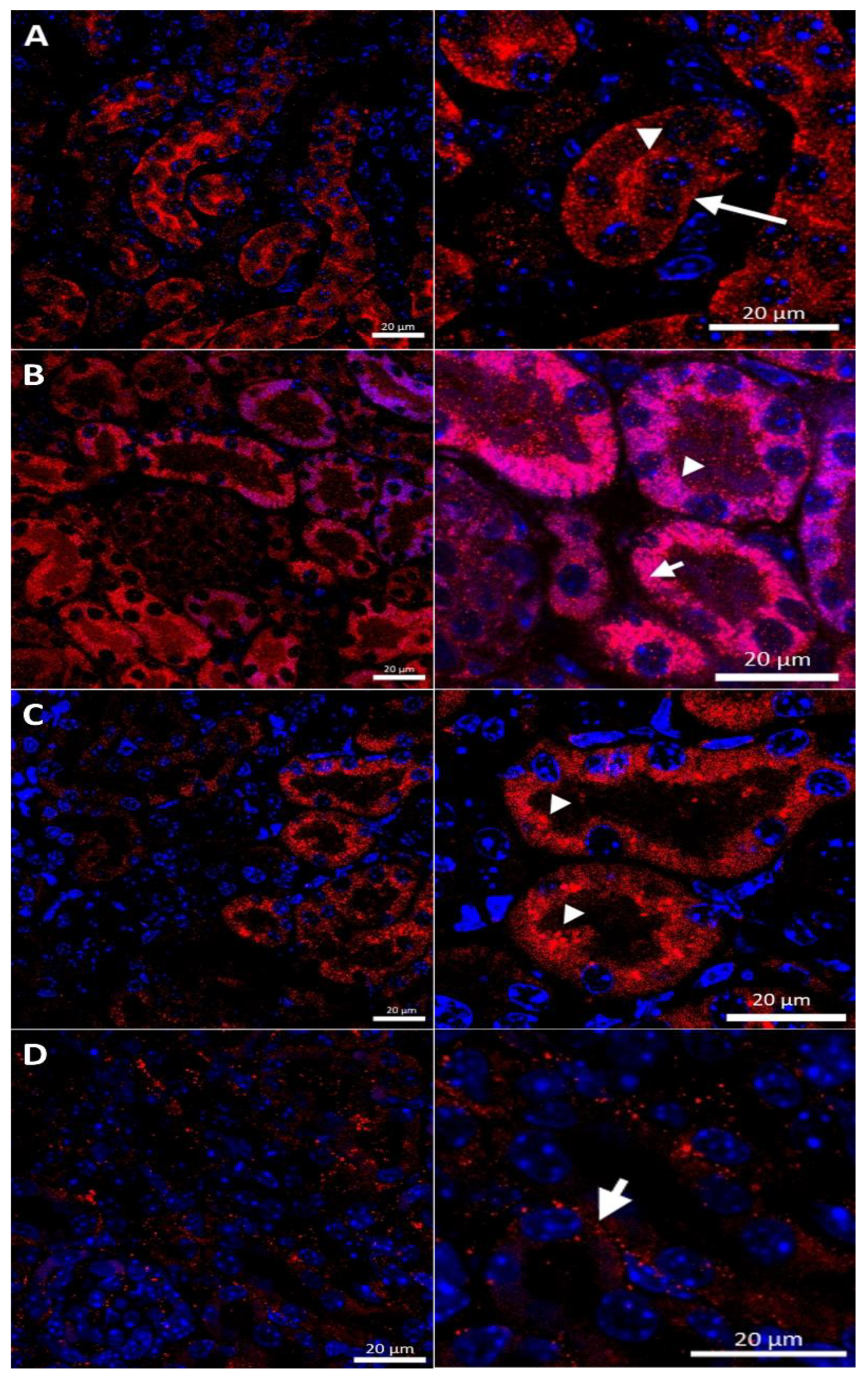
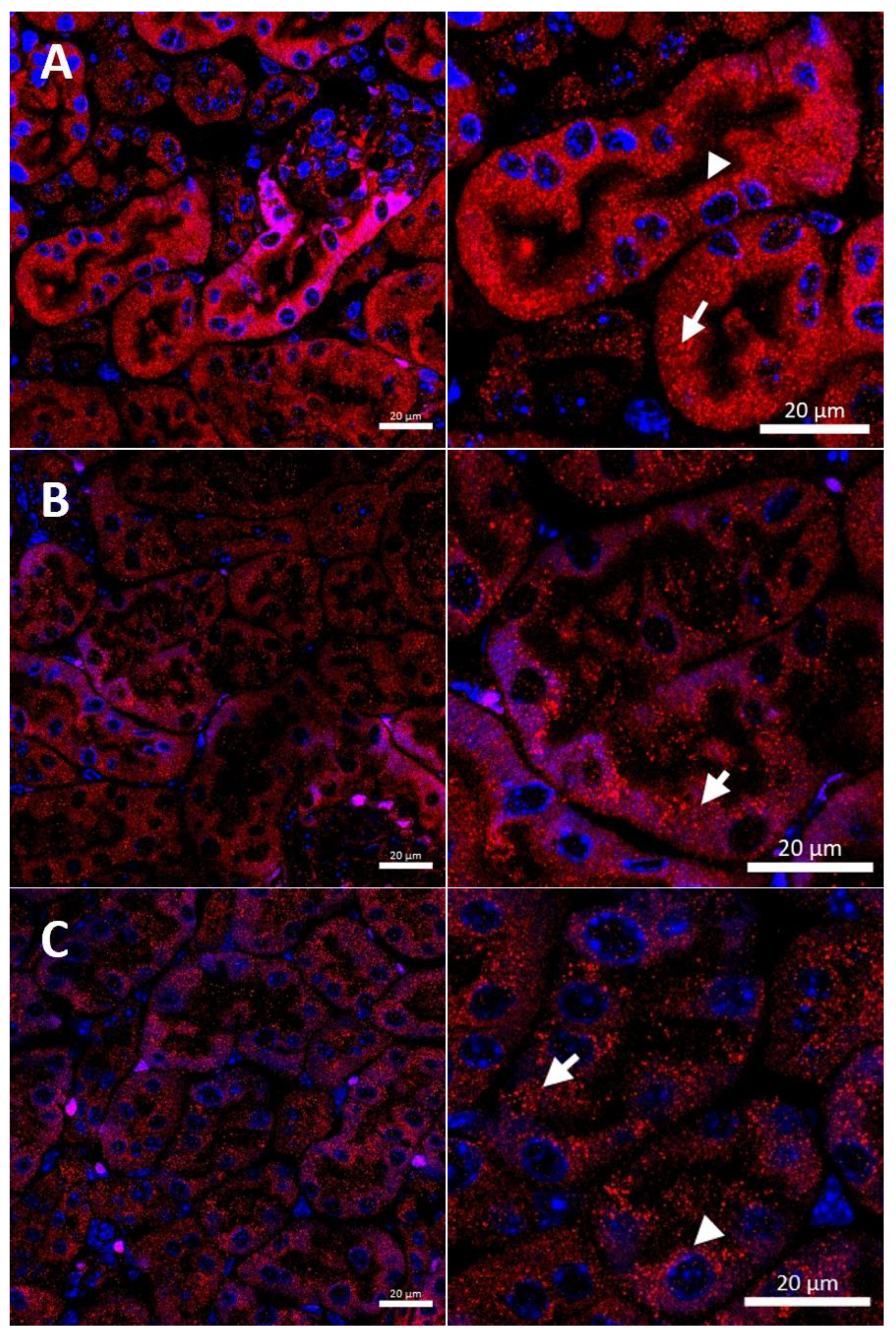
2.3. Effect of Copper Supplementation on The CTR1 Protein Localization in the Kidney of 14-Day-Old Mosaic Mutant and W-T Mice
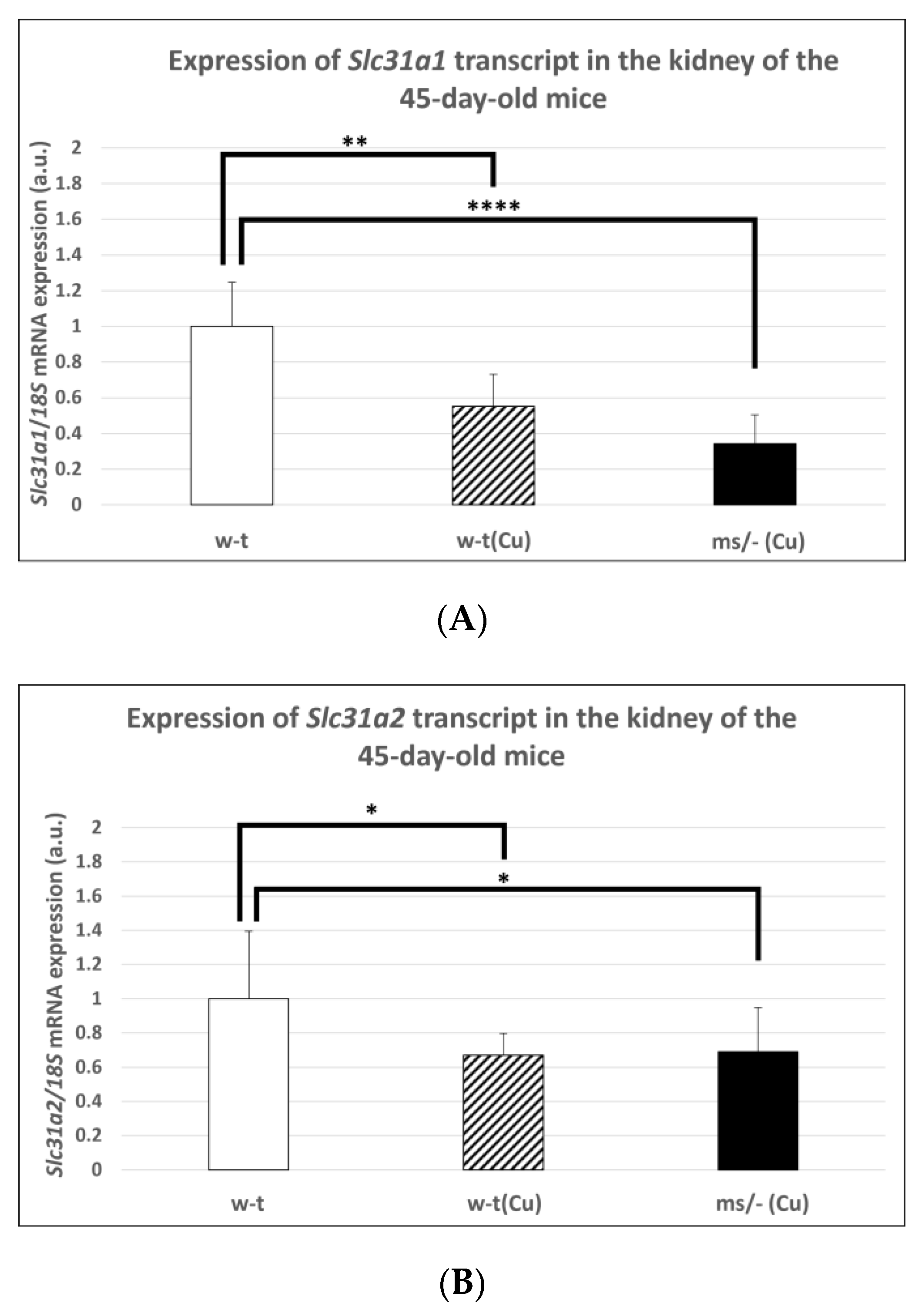
2.4. Effect of the Long-Term Copper Supplementation on the Renal Expression of the Slc31a1 and Slc31a2 Genes at the mRNA Level in 45-Day-Old Mosaic Mutant and W-T Mice
2.5. Effect of the Long-Term Copper Supplementation on the CTR1 Protein Localization in the Kidney of 45-Day-Old Mosaic Mutant and W-T Mice
2.6. Effect of the Long-Term Copper Supplementation on the Renal Expression of The Slc31a1 and Slc31a2 Genes at the mRNA Level in 6-Month-Old Mosaic Mutant and W-T Mice
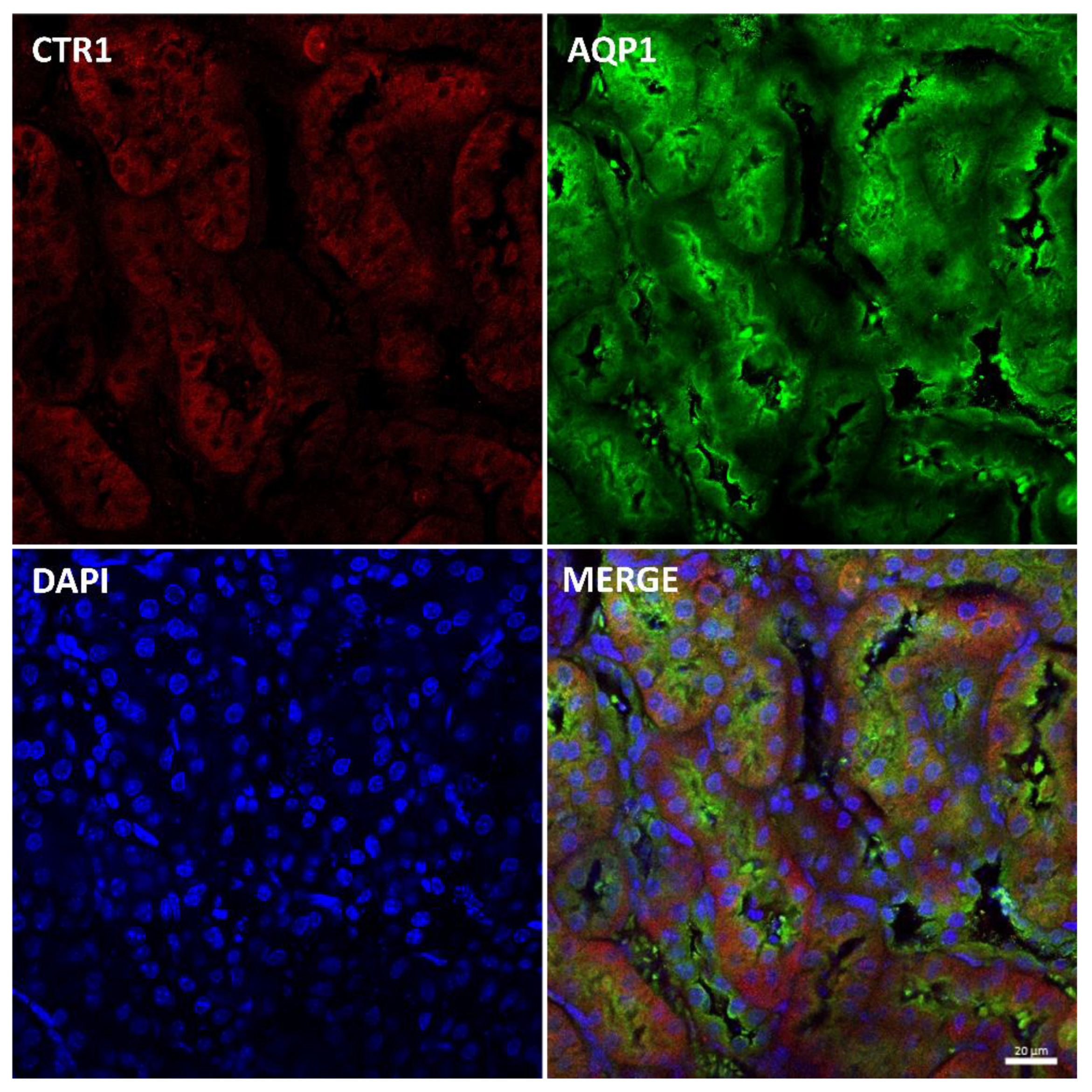
2.7. Effect of the Long-Term Copper Supplementation on the CTR1 Protein Localization in the Kidney of 6-Month-Old W-T and Mosaic Mutant Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Copper Therapy
4.2. Tissue Sample Collection
4.3. Measurement of the Total Copper Content in Tissues
4.4. Real-Time Quantitative RT-PCR
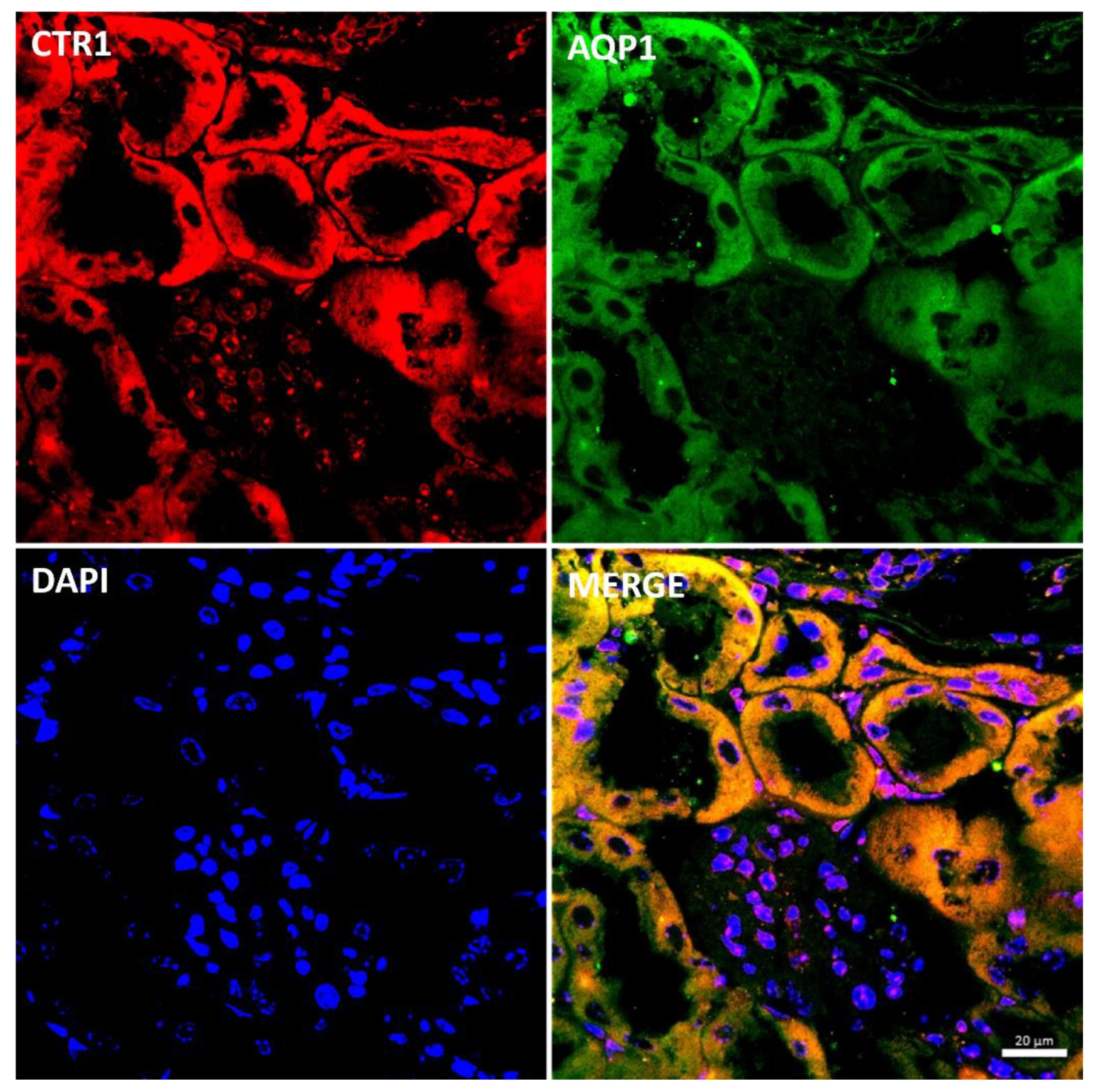
4.5. Immunofluorescence (IF) and Confocal Analysis of Kidney Sections
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Terao, T.; Owen, C.A.J. Copper Metabolism in Pregnant and Pospartum Rat and Pups. Am. J. Physiol. 1977, 232, E172–E179. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, M.; Krzeptowski, W.; Lipinski, P.; Grzmil, P.; Starzynski, R.; Pierzchala, O.; Møller, L.B. Mottled Mice and Non-Mammalian Models of Menkes Disease. Front. Mol. Neurosci. 2015, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and Regulation of Human Copper-Transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited Copper Transport Disorders: Biochemical Mechanisms, Diagnosis, and Treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Wijmenga, C.; Klomp, L.W.J. Molecular Regulation of Copper Excretion in the Liver. Proc. Nutr. Soc. 2004, 63, 31–39. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Windak, R.; Tylko, G.; Kowal, M.; Styrna, J. Effects of Copper Supplementation on the Structure and Content of Elements in Kidneys of Mosaic Mutant Mice. Biol. Trace Elem. Res. 2010, 136, 204–220. [Google Scholar] [CrossRef]
- Linz, R.; Barnes, N.L.; Zimnicka, A.M.; Kaplan, J.H.; Eipper, B.; Lutsenko, S. Intracellular Targeting of Copper-Transporting ATPase ATP7A in a Normal and Atp7b-/- Kidney. Am. J. Physiol. Renal Physiol. 2008, 294, F53–F61. [Google Scholar] [CrossRef]
- Klomp, A.E.M.; Tops, B.B.J.; van Denberg, I.E.T.; Berger, R.; Klomp, L.W.J. Biochemical Characterization and Subcellular Localization of Human Copper Transporter 1 (hCTR1). Biochem. J. 2002, 364, 497–505. [Google Scholar] [CrossRef]
- Öhrvik, H.; Thiele, D.J. The Role of Ctr1 and Ctr2 in Mammalian Copper Homeostasis and Platinum-Based Chemotherapy. J. Trace Elem. Med. Biol. 2015, 31, 178–182. [Google Scholar] [CrossRef]
- Ren, F.; Logeman, B.L.; Zhang, X.; Liu, Y.; Thiele, D.J.; Yuan, P. X-ray Structures of the High-Affinity Copper Transporter Ctr1. Nat. Commun. 2019, 10, 1386. [Google Scholar] [CrossRef] [Green Version]
- Öhrvik, H.; Thiele, D.J. How Copper Traverses Cellular Membranes through the Mammalian Copper Transporter 1, Ctr1. Ann. N. Y. Acad. Sci. 2014, 1314, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Zimnicka, A.M.; Kaplan, J.H. Copper Entry into Human Cells: Progress and Unanswered Questions. Biometals 2007, 20, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Smith, K.; Lee, J.; Thiele, D.J.; Petris, M.J. Identification of Methionine-Rich Clusters That Regulate Copper-Stimulated Endocytosis of the Human Ctr1 Copper Transporter. J. Biol. Chem. 2004, 279, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Öhrvik, H.; Aaseth, J.; Horn, N. Orchestration of Dynamic Copper Navigation—New and Missing Pieces. Metallomics 2017, 9, 1204–1229. [Google Scholar] [CrossRef] [PubMed]
- Nevitt, T.; Öhrvik, H.; Thiele, D.J. Charting the Travels of Copper in Eukaryotes from Yeast to Mammals. Biochim. Biophys. Acta 2012, 1823, 1580–1593. [Google Scholar] [CrossRef] [PubMed]
- Wee, N.K.Y.; Weinstein, D.C.; Fraser, S.T.; Assinder, S.J. The Mammalian Copper Transporters CTR1 and CTR2 and Their Roles in Development and Disease. Int. J. Biochem. Cell Biol. 2013, 45, 960–963. [Google Scholar] [CrossRef]
- Zimnicka, A.M.; Maryon, E.B.; Kaplan, J.H. Human Copper Transporter hCTR1 Mediates Basolateral Uptake of Copper into Enterocytes: Implications for Copper Homeostasis. J. Biol. Chem. 2007, 282, 26471–26480. [Google Scholar] [CrossRef]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The Copper Transporter Ctr1 Contributes to Cisplatin Uptake by Renal Tubular Cells during Cisplatin Nephrotoxicity. Am. J. Physiol. Renal Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Gybina, A.A.; Pyatskowit, J.W.; Gitschier, J.; Prohaska, J.R. Copper Transport Protein (Ctr1) Levels in Mice Are Tissue Specific and Dependent on Copper Status. J. Nutr. 2006, 136, 21–26. [Google Scholar] [CrossRef]
- Eisses, J.F.; Chi, Y.; Kaplan, J.H. Stable Plasma Membrane Levels of hCTR1 Mediate Cellular Copper Uptake. J. Biol. Chem. 2005, 280, 9635–9639. [Google Scholar] [CrossRef] [Green Version]
- Clifford, R.J.; Maryon, E.B.; Kaplan, J.H. Dynamic Internalization and Recycling of a Metal Ion Transporter: Cu Homeostasis and CTR1, the Human Cu+ Uptake System. J. Cell Sci. 2016, 129, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Tümer, Z.; Møller, L.B. Menkes Disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Tümer, Z. An Overview and Update of ATP7A Mutations Leading to Menkes Disease and Occipital Horn Syndrome. Hum. Mutat. 2013, 34, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G.; Holmes, C.S.; Goldstein, D.S.; Tang, J.; Godwin, S.C.; Donsante, A.; Liew, C.J.; Sato, S.; Patronas, N. Neonatal Diagnosis and Treatment of Menkes Disease. N. Engl. J. Med. 2008, 358, 605–614. [Google Scholar] [CrossRef]
- Møller, L.B.; Mogensen, M.; Horn, N. Molecular Diagnosis of Menkes Disease: Genotype-Phenotype Correlation. Biochimie 2009, 91, 1273–1277. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Grzmil, P.; Shoukier, M.; Starzynski, R.; Marciniak, M.; Lipinski, P. Mutation in the CPC Motif-Containing 6th Transmembrane Domain Affects Intracellular Localization, Trafficking and Copper Transport Efficiency of ATP7A Protein in Mosaic Mutant Mice-an Animal Model of Menkes Disease. Metallomics 2012, 4, 197–204. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Krzeptowski, W.; Koteja, P.; Chrząścik, K.; Møller, L.B. Prenatal Treatment of Mosaic Mice (Atp7a Mo-Ms) Mouse Model for Menkes Disease, with Copper Combined by Dimethyldithiocarbamate (DMDTC). PLoS ONE 2012, 7, e40400. [Google Scholar] [CrossRef]
- Horn, N.; Møller, L.B.; Nurchi, V.M.; Aaseth, J. Chelating Principles in Menkes and Wilson Diseases: Choosing the Right Compounds in the Right Combinations at the Right Time. J. Inorg. Biochem. 2019, 190, 98–112. [Google Scholar] [CrossRef]
- la Fontaine, S.; Mercer, J.F.B. Trafficking of the Copper-ATPases, ATP7A and ATP7B: Role in Copper Homeostasis. Arch. Biochem. Biophys. 2007, 463, 149–167. [Google Scholar] [CrossRef]
- el Meskini, R.; Culotta, V.C.; Mains, R.E.; Eipper, B.A. Supplying Copper to the Cuproenzyme Peptidylglycine Alpha-Amidating Monooxygenase. J. Biol. Chem. 2003, 278, 12278–12284. [Google Scholar] [CrossRef] [Green Version]
- Proud, V.K.; Mussell, H.G.; Kaler, S.G.; Young, D.W.; Percy, A.K. Distinctive Menkes Disease Variant with Occipital Horns: Delineation of Natural History and Clinical Phenotype. Am. J. Med. Genet. 1996, 65, 44–51. [Google Scholar] [CrossRef]
- Kodama, H.; Gu, Y.-H.; Mizunuma, M. Drug Targets in Menkes Disease—Prospective Developments. Expert Opin. Ther. Targets 2001, 5, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Chong, P.F.; Maeda, K.; Imagi, T.; Nakamura, R.; Kawamura, N.; Kira, R. Long Surviving Classical Menkes Disease Treated with Weekly Intravenous Copper Therapy. J. Trace Elem. Med. Biol. 2019, 54, 172–174. [Google Scholar] [CrossRef]
- Oshio, T.; Hino, M.; Kirino, A.; Matsumura, C.; Fukuda, K. Urologic Abnormalities in Menkes’ Kinky Hair Disease: Report of Three Cases. J. Pediatr. Surg. 1997, 32, 782–784. [Google Scholar] [CrossRef]
- ClinicalTrials.gov NIH U.S. National Library of Medicine; Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency. Available online: https://clinicaltrials.gov/ct2/show/NCT00811785 (accessed on 28 August 2020).
- Levinson, B.; Vulpe, C.; Elder, B.; Martin, C.; Verley, F.; Packman, S.; Gitschier, J. The Mottled Gene Is the Mouse Homologue of the Menkes Disease Gene. Nat. Genet. 1994, 6, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Biasotto, M.; Tosi, M.; Avner, P. The Mottled Mouse as a Model for Human Menkes Disease: Identification of Mutations in the Atp7a Gene. Hum. Mol. Genet. 1997, 6, 425–433. [Google Scholar] [CrossRef]
- la Fontaine, S.; Firth, S.D.; Lockhart, P.J.; Brooks, H.; Camakaris, J.; Mercer, J.F. Intracellular Localization and Loss of Copper Responsiveness of Mnk, the Murine Homologue of the Menkes Protein, in Cells from Blotchy (Mo Blo) and Brindled (Mo Br) Mouse Mutants. Hum. Mol. Genet. 1999, 8, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Horiike, K.; Nozaki, M.; Shimada, M. Copper metabolism in the macular mouse, an animal model for Menke’s kinky hair disease: Changes in the cytochrome C oxidase activity and copper content in various organs after copper administration. Biomed. Res. 1993, 14, 57–64. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Starzyński, R.R.; Jończy, A.; Staroń, R.; Antoniuk, J.; Krzeptowski, W.; Grzmil, P.; Bednarz, A.; Pierzchała, O.; Ogórek, M.; et al. Copper Therapy Reduces Intravascular Hemolysis and Derepresses Ferroportin in Mice with Mosaic Mutation (Atp7a(Mo-Ms)): An Implication for Copper-Mediated Regulation of the Slc40a1 Gene Expression. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1410–1421. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Sasuła, K. Altered copper metabolism in the mosaic mutant mice. Nutr. Res. 2000, 10, 1519–1529. [Google Scholar] [CrossRef]
- Kowal, M.; Lenartowicz, M.; Pecio, A.; Golas, A.; Blaszkiewicz, T.; Styrna, J. Copper Metabolism Disorders Affect Testes Structure and Gamete Quality in Male Mice. Syst. Biol. Reprod. Med. 2010, 56, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Styrna, J. Analysis of causes of lethality in mice with Ms (Mosaic) gene. Genet. Polon 1977, 18, 61–79. [Google Scholar]
- Liang, Z.D.; Tsai, W.-B.; Lee, M.-Y.; Savaraj, N.; Kuo, M.T. Specificity Protein 1 (Sp1) Oscillation Is Involved in Copper Homeostasis Maintenance by Regulating Human High-Affinity Copper Transporter 1 Expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Starzyński, R.R.; Krzeptowski, W.; Grzmil, P.; Bednarz, A.; Ogórek, M.; Pierzchała, O.; Staroń, R.; Gajowiak, A.; Lipiński, P. Haemolysis and Perturbations in the Systemic Iron Metabolism of Suckling, Copper-Deficient Mosaic Mutant Mice—An Animal Model of Menkes Disease. PLoS ONE 2014, 9, e107641. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Long, Y.; Chen, H.H.W.; Savaraj, N.; Kuo, M.T. Regulation of the High-Affinity Copper Transporter (hCtr1) Expression by Cisplatin and Heavy Metals. J. Biol. Inorg. Chem. 2014, 19, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Chen, H.H.W.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.J.; Kuo, M.T. Transcription Factor Sp1 Plays an Important Role in the Regulation of Copper Homeostasis in Mammalian Cells. Mol. Pharmacol. 2008, 74, 705–713. [Google Scholar] [CrossRef]
- Molloy, S.A.; Kaplan, J.H. Copper-Dependent Recycling of hCTR1, the Human High Affinity Copper Transporter. J. Biol. Chem. 2009, 284, 29704–29713. [Google Scholar] [CrossRef]
- Smeets, B.; Boor, P.; Dijkman, H.; Sharma, S.V.; Jirak, P.; Mooren, F.; Berger, K.; Bornemann, J.; Gelman, I.H.; Floege, J.; et al. Proximal Tubular Cells Contain a Phenotypically Distinct, Scattered Cell Population Involved in Tubular Regeneration. J. Pathol. 2013, 229, 645–659. [Google Scholar] [CrossRef]
- Nielsen, S.; Frøkiaer, J.; Marples, D.; Kwon, T.-H.; Agre, P.; Knepper, M.A. Aquaporins in the Kidney: From Molecules to Medicine. Physiol. Rev. 2002, 82, 205–244. [Google Scholar] [CrossRef]
- Petris, M.J.; Smith, K.; Lee, J.; Thiele, D.J. Copper-Stimulated Endocytosis and Degradation of the Human Copper Transporter, hCtr1. J. Biol. Chem. 2003, 278, 9639–9646. [Google Scholar] [CrossRef]
- Nose, Y.; Wood, L.K.; Kim, B.E.; Prohaska, J.R.; Fry, R.S.; Spears, J.W.; Thiele, D.J. Ctr1 Is an Apical Copper Transporter in Mammalian Intestinal Epithelial Cells in Vivo That Is Controlled at the Level of Protein Stability. J. Biol. Chem. 2010, 285, 32385–32392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenartowicz, M.; Kowal, M.; Buda-Lewandowska, D.; Styrna, J. Pathological Structure of the Kidney from Adult Mice with Mosaic Mutation. J. Inherit. Metab. Dis. 2002, 25, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.J.; Danks, D.M.; Legge, G.J.; Mercer, J.F. Analysis of the Distribution of Cu, Fe and Zn and Other Elements in Brindled Mouse Kidney Using a Scanning Proton Microprobe. J. Inorg. Biochem. 1998, 71, 189–197. [Google Scholar] [CrossRef]
- Suzuki-Kurasaki, M.; Okabe, M.; Kurasaki, M. Copper-Metallothionein in the Kidney of Macular Mice: A Model for Menkes Disease. J. Histochem. Cytochem. 1997, 45, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Nooijen, J.L.; de Groot, C.J.; van den Hamer, C.J.; Monnens, L.A.; Willemse, J.; Niermeijer, M.F. Trace Element Studies in Three Patients and a Fetus with Menkes’ Disease. Effect of Copper Therapy. Pediatr. Res. 1981, 15, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Zaffanello, M.; Maffeis, C.; Fanos, V.; Franchini, M.; Zamboni, G. Urological Complications and Copper Replacement Therapy in Childhood Menkes Syndrome. Acta Paediatr. 2006, 95, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Kim, J.H.; Cho, M.H.; Choi, Y.H.; Kim, S.H.; Im, Y.J.; Park, K.; Kang, H.G.; Chae, J.-H.; Cheong, H. il Urological Problems in Patients with Menkes Disease. J. Korean Med. Sci. 2019, 34, e4. [Google Scholar] [CrossRef]
- Horn, N.; Heydorn, K.; Damsgaard, E.; Tygstrup, I.; Vestermark, S. Is Menkes Syndrome a Copper Storage Disorder? Clin. Genet. 1978, 14, 186–187. [Google Scholar] [CrossRef]
- Kodama, H.; Sato, E.; Gu, Y.-H.; Shiga, K.; Fujisawa, C.; Kozuma, T. Effect of Copper and Diethyldithiocarbamate Combination Therapy on the Macular Mouse, an Animal Model of Menkes Disease. J. Inherit. Metab. Dis. 2005, 28, 971–978. [Google Scholar] [CrossRef]
- Prohaska, J.R. Changes in Tissue Growth, Concentrations of Copper, Iron, Cytochrome Oxidase and Superoxide Dismutase Subsequent to Dietary or Genetic Copper Deficiency in Mice. J. Nutr. 1983, 113, 2048–2058. [Google Scholar] [CrossRef]
- Rees, E.M.; Lee, J.; Thiele, D.J. Mobilization of Intracellular Copper Stores by the Ctr2 Vacuolar Copper Transporter. J. Biol. Chem. 2004, 279, 54221–54229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.D.; Chan, J.; et al. Wilson Disease Protein ATP7B Utilizes Lysosomal Exocytosis to Maintain Copper Homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.H.W. Role of the Human High-Affinity Copper Transporter in Copper Homeostasis Regulation and Cisplatin Sensitivity in Cancer Chemotherapy. Cancer Res. 2012, 72, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, P.V.E.; Folmer, D.E.; Malingré, H.E.M.; van Beurden, E.; Klomp, A.E.M.; van de Sluis, B.; Merkx, M.; Berger, R.; Klomp, L.W.J. Human Copper Transporter 2 Is Localized in Late Endosomes and Lysosomes and Facilitates Cellular Copper Uptake. Biochem. J. 2007, 407, 49–59. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age/Genotype | Copper Concentration [μg/g Wet Tissue] | ||
|---|---|---|---|
| Liver | Kidney | Urine | |
| 14-day-old w-t (5) | 8.01 ± 2.11 | 2.06 ± 0.22 | 0.08 ± 0.03 |
| 14-day-old w-t (Cu) (5) | 67.40 ± 2644 a **, b ** | 3.13 ± 1.21 b **** | 0.03 ± 0.02 |
| 14-day-old ms/− (5) | 2.49 ± 0.26 a **, b ** | 6.31 ± 1.36 b ***, c *** | N.A. |
| 14-day-old ms/− (Cu) (5) | 3.80 ± 0.58 a **, c ***, d ** | 24.58 ± 828 a ****, c ***, d **** | 0.29 ± 0.13 a *, d * |
| 45-day-old w-t (5) | 5.31 ± 0.98 | 3.42 ± 0.61 | 0.21 ± 0.13 |
| 45-day-old w-t (Cu) (5) | 4.45 ± 1.26 | 3.81 ± 0.39 b **** | 0.16 ± 0.07 |
| 45-day-old ms/− (Cu) (5) | 3.41 ± 0.52 a * | 4051 ± 10.12 a ****, d **** | 0.23 ± 0.193 |
| 6-month-old w-t (5) | 4.17 ± 0.84 | 4.40 ± 1.00 | 0.15 ± 0.05 |
| 6-month-old w-t (Cu) (5) | 4.65 ± 0.69 | 3.70 ± 0.60 b **** | 0.19 ± 0.11 |
| 6-month-old ms/− (Cu) (5) | 3.44 ± 0.34 a * | 22.70 ± 5.20 a ****, d **** | 0.17 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haberkiewicz, O.; Lipiński, P.; Starzyński, R.R.; Jończy, A.; Kurowska, P.; Ogórek, M.; Bednarz, A.; Herman, S.; Hatala, D.; Grzmil, P.; et al. Decreased Expression of the Slc31a1 Gene and Cytoplasmic Relocalization of Membrane CTR1 Protein in Renal Epithelial Cells: A Potent Protective Mechanism against Copper Nephrotoxicity in a Mouse Model of Menkes Disease. Int. J. Mol. Sci. 2022, 23, 11441. https://doi.org/10.3390/ijms231911441
Haberkiewicz O, Lipiński P, Starzyński RR, Jończy A, Kurowska P, Ogórek M, Bednarz A, Herman S, Hatala D, Grzmil P, et al. Decreased Expression of the Slc31a1 Gene and Cytoplasmic Relocalization of Membrane CTR1 Protein in Renal Epithelial Cells: A Potent Protective Mechanism against Copper Nephrotoxicity in a Mouse Model of Menkes Disease. International Journal of Molecular Sciences. 2022; 23(19):11441. https://doi.org/10.3390/ijms231911441
Chicago/Turabian StyleHaberkiewicz, Olga, Paweł Lipiński, Rafał R. Starzyński, Aneta Jończy, Patrycja Kurowska, Mateusz Ogórek, Aleksandra Bednarz, Sylwia Herman, Dawid Hatala, Paweł Grzmil, and et al. 2022. "Decreased Expression of the Slc31a1 Gene and Cytoplasmic Relocalization of Membrane CTR1 Protein in Renal Epithelial Cells: A Potent Protective Mechanism against Copper Nephrotoxicity in a Mouse Model of Menkes Disease" International Journal of Molecular Sciences 23, no. 19: 11441. https://doi.org/10.3390/ijms231911441