

Gut–Brain Axis: Insights from Hippocampal Neurogenesis and Brain Tumor Development in a Mouse Model of Experimental Colitis Induced by Dextran Sodium Sulfate
 ,
,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
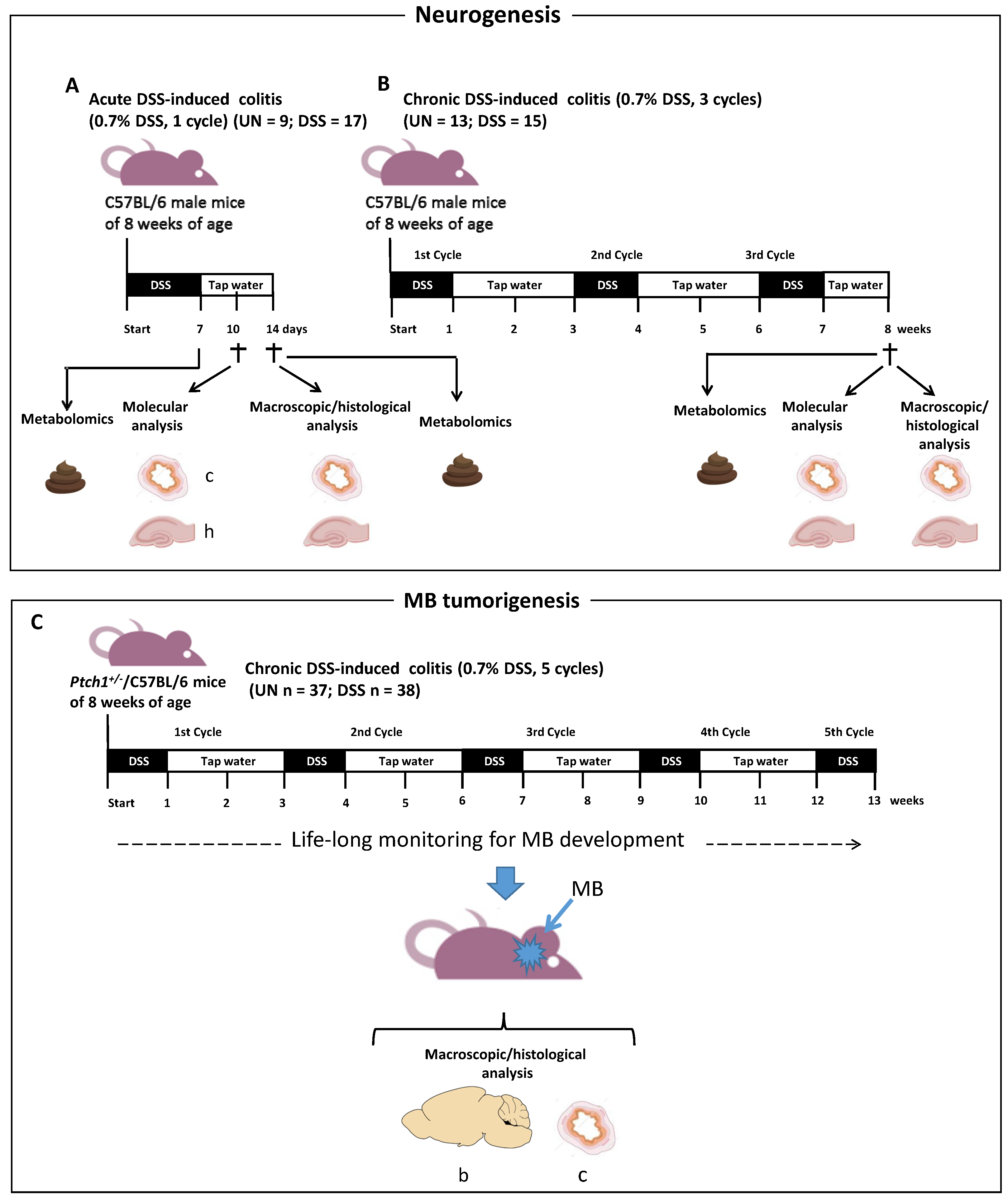
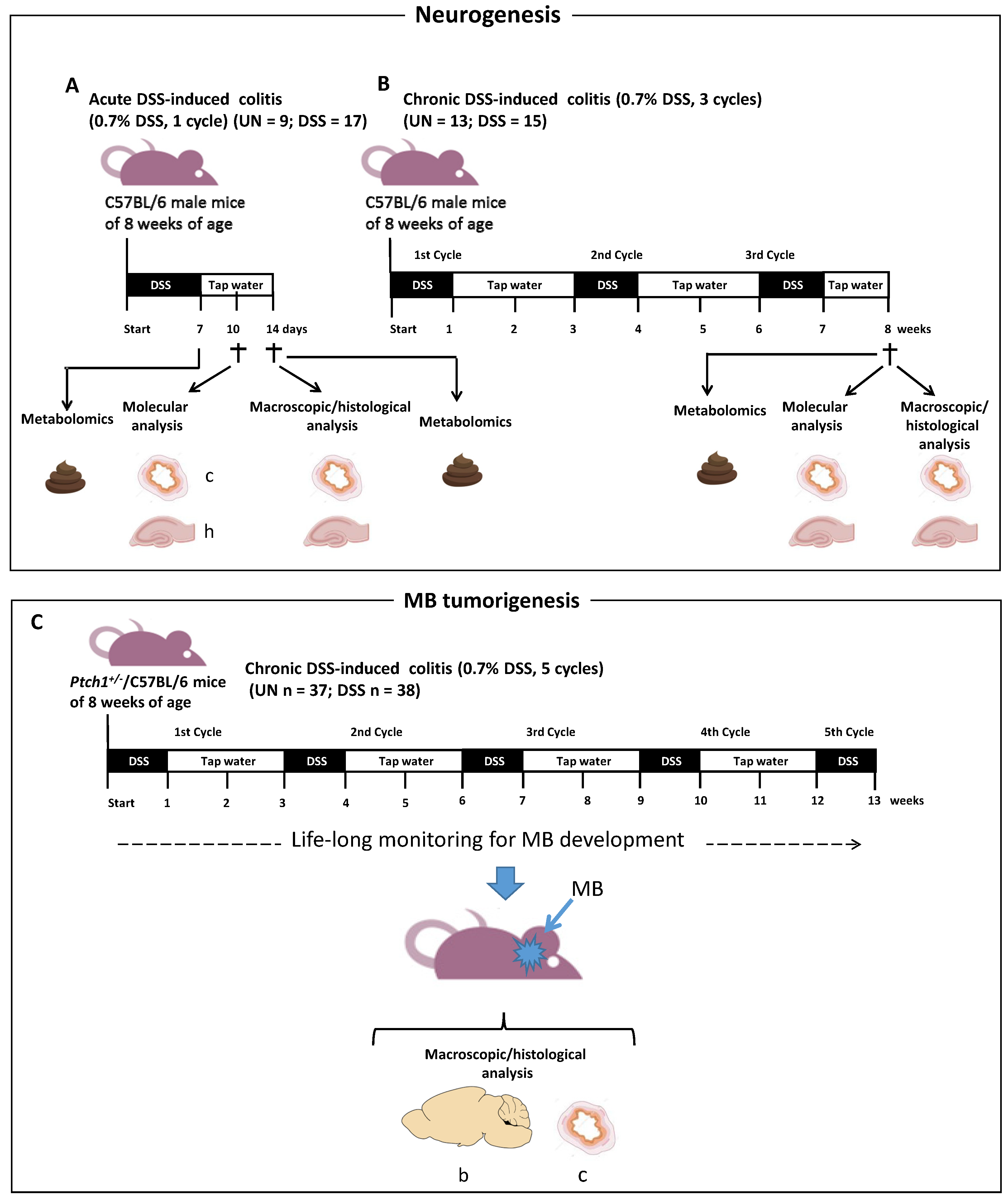
2.1. Schematic Experimental Design
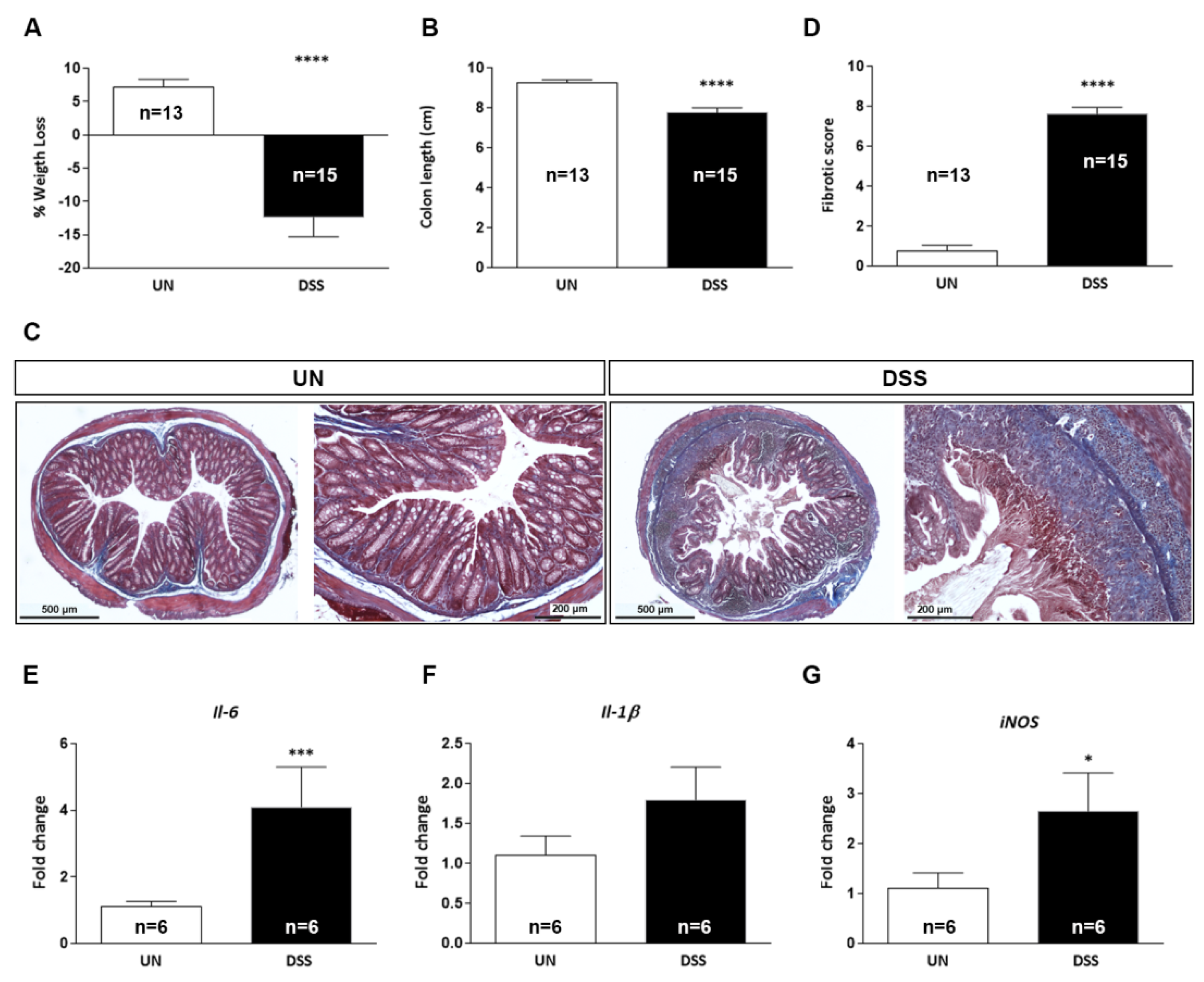
2.2. Characterization of Acute DSS-Induced Colitis
2.3. Characterization of Chronic DSS-Induced Colitis
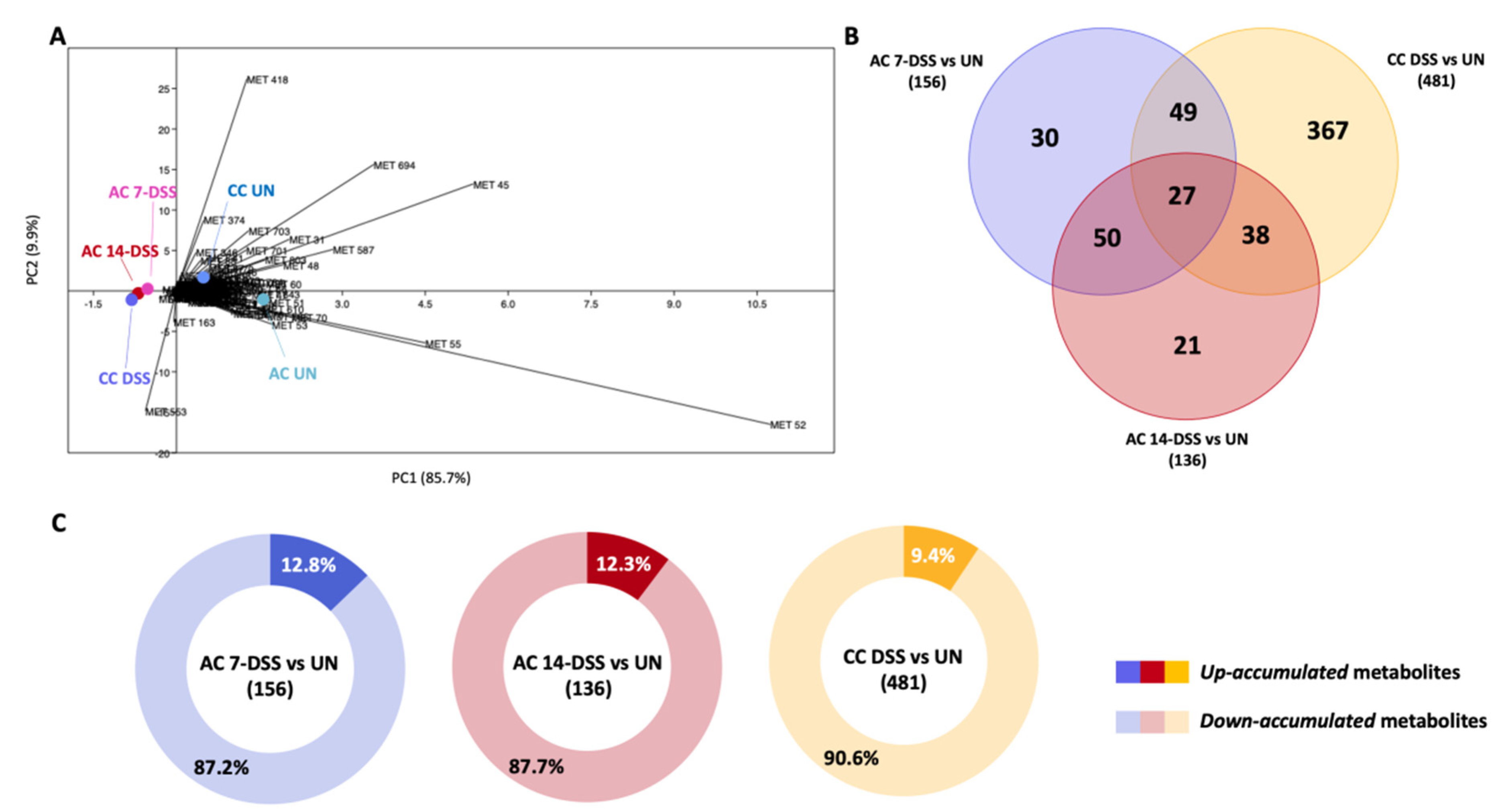
2.4. Comparative Study of the Metabolomes in Fecal Samples from DSS-Treated and Untreated Mice
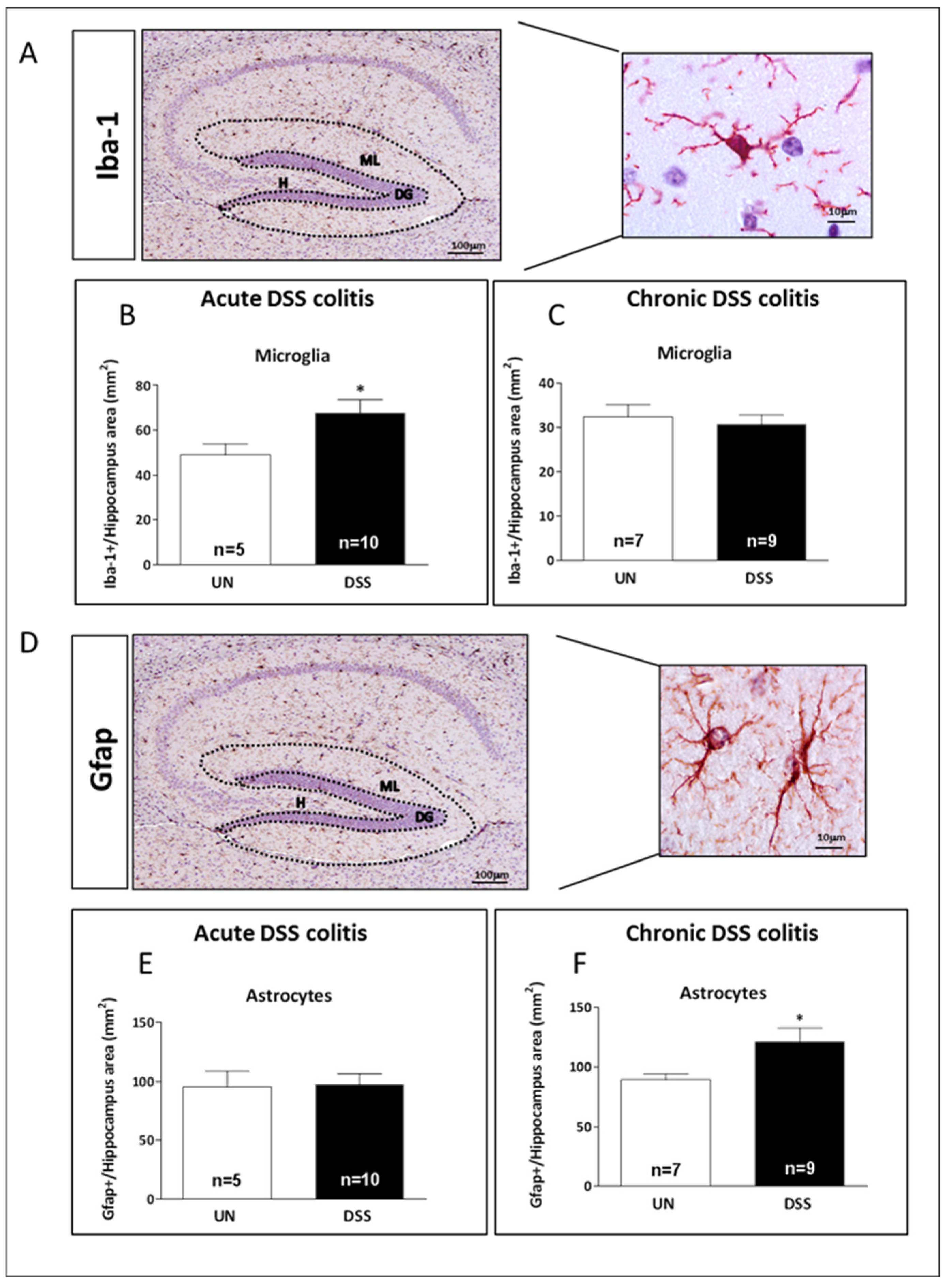
2.5. Acute and Chronic Colitis Enhanced Inflammatory Response in the Hippocampus with a Distinct Cellular Pattern
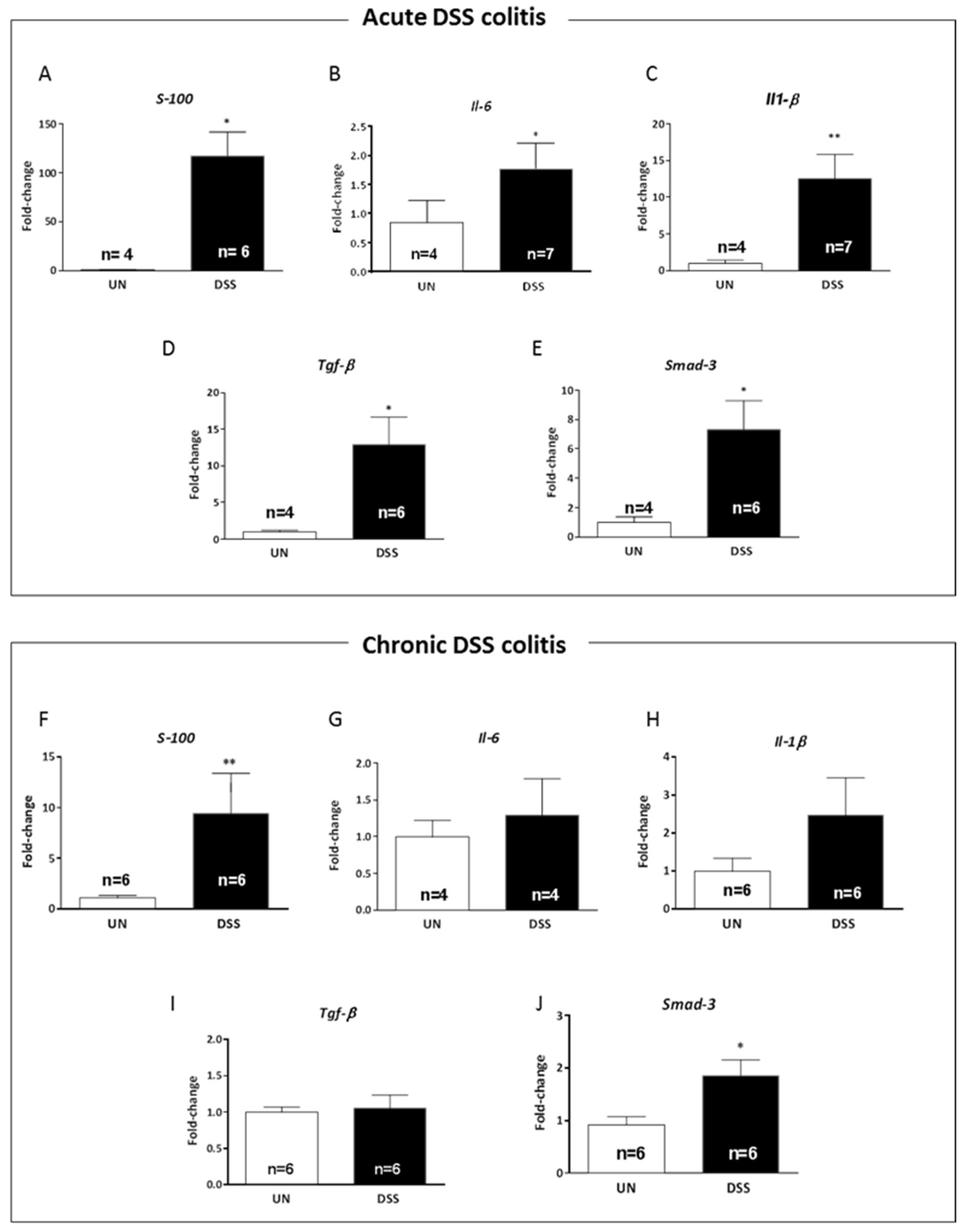
2.6. Acute and Chronic Colitis Enhanced Inflammatory Response in the Hippocampus with a Distinct Cytokine Profile
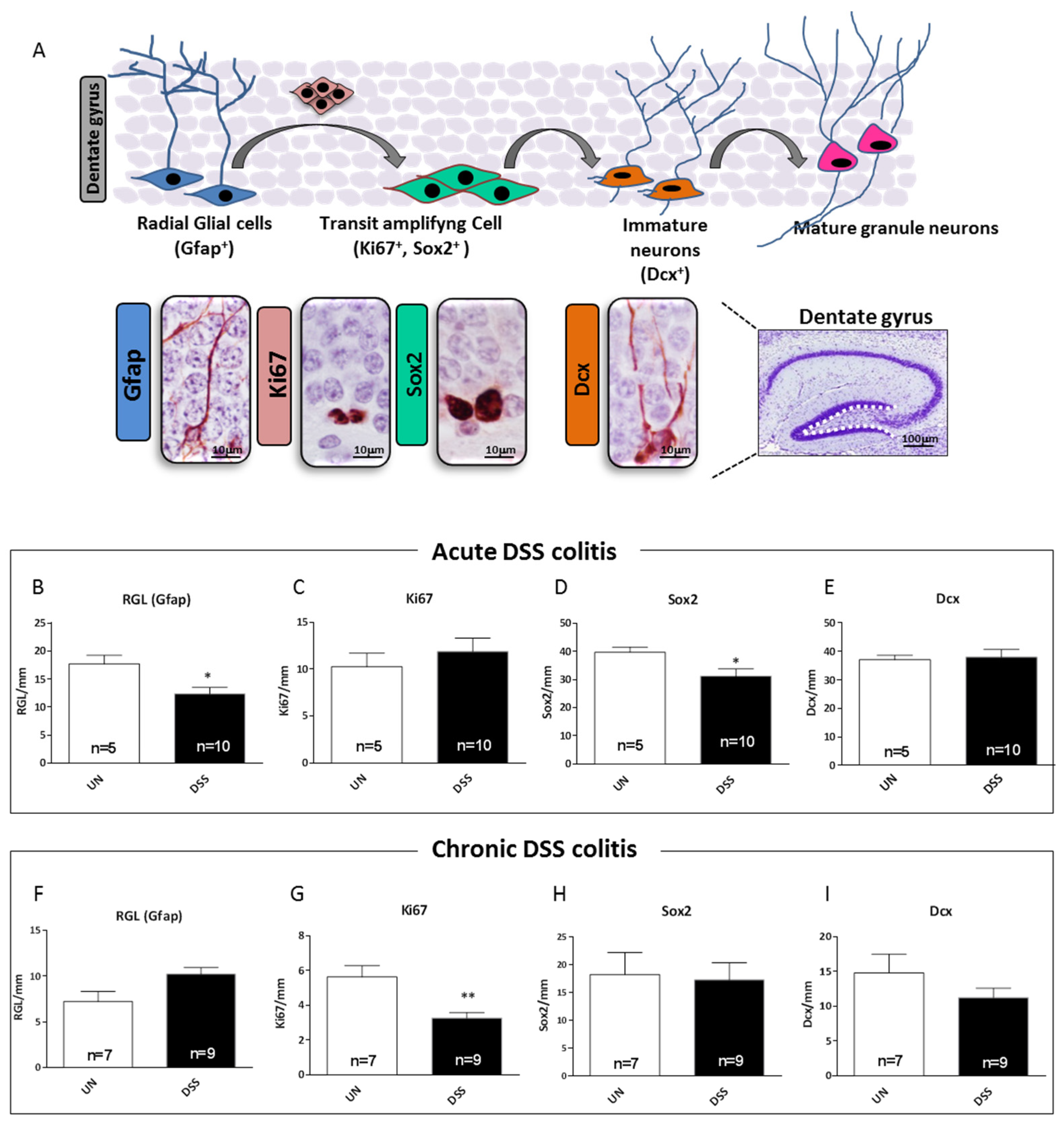
2.7. Acute and Chronic Colitis Induced Different Stage-Specific Changes in Hippocampal Neurogenesis
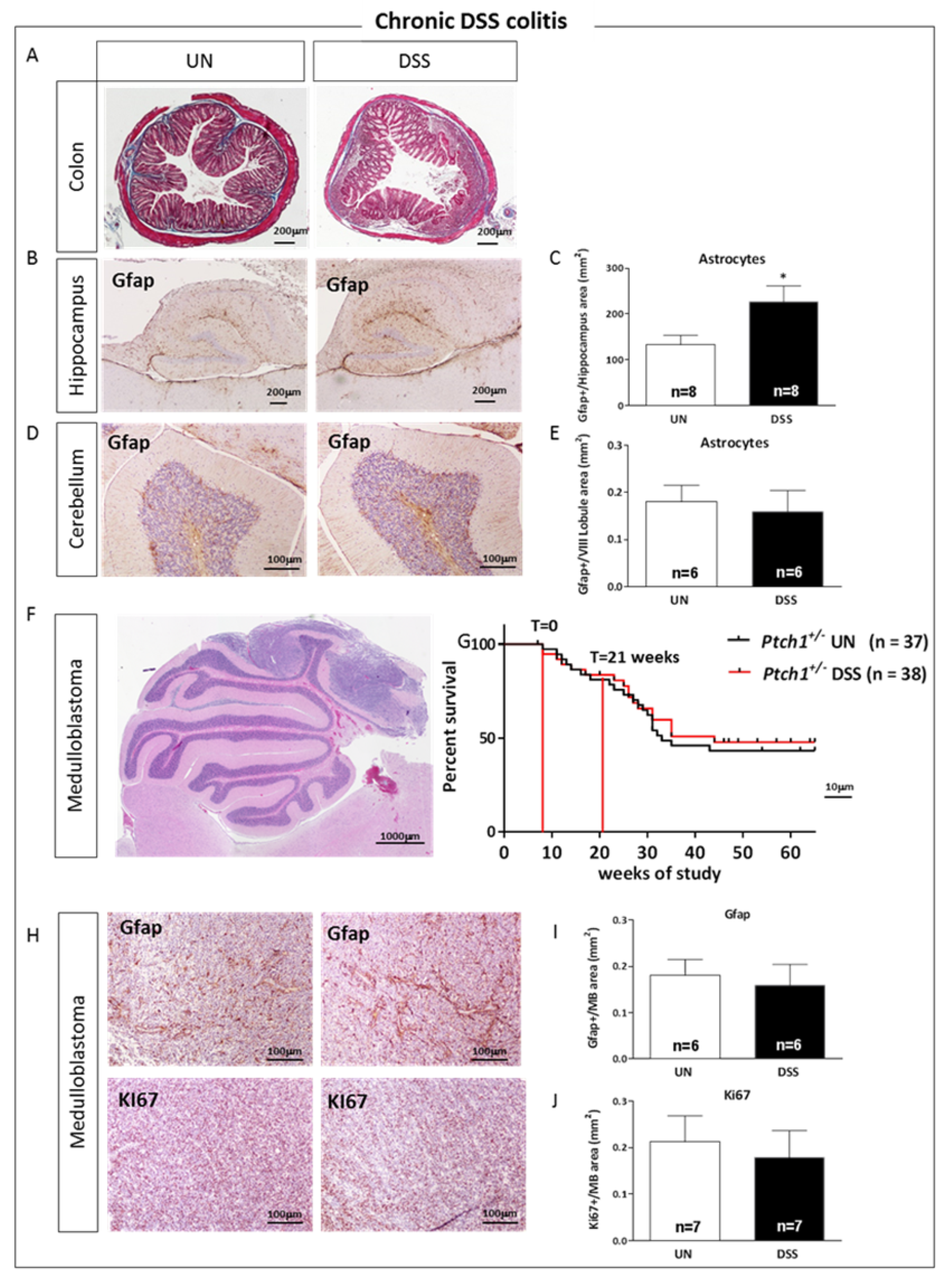
2.8. Relationship between Gut Inflammation and Brain Tumorigenesis in Ptch1+/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Treatment with Dextran Sodium Sulfate (DSS)
4.3. Assessment of Acute and Chronic DSS-Induced Colitis
4.4. Collection of Brain Samples
4.5. Immunohistochemistry
4.6. Real-Time qPCR
4.7. Processing of Stool Samples and Metabolomics
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BBB | Brain–blood barrier |
| CNS | Central nervous system |
| DAMs | Differentially accumulated metabolites |
| Dcx | Doublecortin |
| DG | Dentate gyrus |
| DSS | Dextran sulfate sodium |
| EGL | External granular layer |
| Gfap | Glial fibrillary acidic protein |
| H | Hilus |
| Iba1 | Ionized calcium-binding adapter molecule 1 |
| IBD | Inflammatory bowel disease |
| iNOS | Inducible nitric oxide |
| IL | Interleukin |
| MB | Medulloblastoma |
| ML | Molecular layer |
| NPC | Neural progenitor cells |
| NSC | Neural stem cells |
| RGL | Radial glia-like |
| SGZ | Sub granular zone |
| Sox2 | SRY (sex determining region Y)-box 2 |
| Tgf | Transforming growth factor |
References
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Chang, L.; Chang, H.M.; Chang, F. Intestinal and Extraintestinal Cancers Associated with Inflammatory Bowel Disease. Clin. Color. Cancer 2018, 17, e29–e37. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Park, A.J.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.A.; Fahnestock, M.; Moine, D.; et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef]
- Zonis, S.; Pechnick, R.N.; Ljubimov, V.A.; Mahgerefteh, M.; Wawrowsky, K.; Michelsen, K.S.; Chesnokova, V. Chronic intestinal inflammation alters hippocampal neurogenesis. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Nakagawasai, O.; Yamada, K.; Takahashi, K.; Odaira, T.; Sakuma, W.; Ishizawa, D.; Takahashi, N.; Onuma, K.; Hozumi, C.; Nemoto, W.; et al. Liver hydrolysate prevents depressive-like behavior in an animal model of colitis: Involvement of hippocampal neurogenesis via the AMPK/BDNF pathway. Behav. Brain Res. 2020, 390, 112640. [Google Scholar] [CrossRef]
- Salvo, E.; Stokes, P.; Keogh, C.E.; Brust-Mascher, I.; Hennessey, C.; Knotts, T.A.; Sladek, J.A.; Rude, K.M.; Swedek, M.; Rabasa, G.; et al. A murine model of pediatric inflammatory bowel disease causes microbiota-gut-brain axis deficits in adulthood. Am. J. Physiol. Liver Physiol. 2020, 319, G361–G374. [Google Scholar] [CrossRef]
- Gampierakis, I.A.; Koutmani, Y.; Semitekolou, M.; Morianos, I.; Polissidis, A.; Katsouda, A.; Charalampopoulos, I.; Xanthou, G.; Gravanis, A.; Karalis, K.P. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Mol. Psychiatry 2020, 26, 1248–1263. [Google Scholar] [CrossRef]
- Ding, Y.; Yanagi, K.; Cheng, C.; Alaniz, R.C.; Lee, K.; Jayaraman, A. Interactions between gut microbiota and non-alcoholic liver disease: The role of microbiota-derived metabolites. Pharmacol. Res. 2019, 141, 521–529. [Google Scholar] [CrossRef]
- Aldars-García, L.; Gisbert, J.P.; Chaparro, M. Metabolomics Insights into Inflammatory Bowel Disease: A Comprehensive Review. Pharmaceuticals 2021, 14, 1190. [Google Scholar] [CrossRef] [PubMed]
- Sitkin, S.I.; Tkachenko, E.I.; Vakhitov, T.Y. Metabolic dysbiosis of the gut microbiota and its. Exp. Clin. Gastroenterol. 2016, 12, 6–29. [Google Scholar]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Viney, J.L. Methods of Inducing Inflammatory Bowel Disease in Mice. Curr. Protoc. Pharmacol. 2009, 47, 5–58. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Scott, M.P. A mouse model for medulloblastoma and basal cell nevus syndrome. J. Neuro-Oncol. 2001, 53, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Marino, S. Medulloblastoma: Developmental mechanisms out of control. Trends Mol. Med. 2005, 11, 17–22. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Pasquali, E.; Tanori, M.; Mancuso, M.; Leonardi, S.; di Majo, V.; Rebessi, S.; Saran, A. Physical heritable and age-related factors as modifiers of radiation cancer risk in patched heterozygous mice. Int. J. Radiat. Oncol. 2009, 73, 1203–1210. [Google Scholar] [CrossRef]
- Mikocka-Walus, A.A.; Turnbull, D.A.; Moulding, N.T.; Wilson, I.G.; Andrews, J.M.; Holtmann, G.J. Antidepressants and inflammatory bowel disease: A systematic review. Clin. Pract. Epidemiol. Ment. Health 2006, 2, 24. [Google Scholar] [CrossRef]
- Graff, L.A.; Walker, J.R.; Bernstein, C.N. Depression and anxiety in inflammatory bowel disease: A review of comorbidity and management. Inflamm. Bowel Dis. 2009, 15, 1105–1118. [Google Scholar] [CrossRef]
- Lydiard, R.B. Irritable bowel syndrome, anxiety, and depression: What are the links? J. Clin. Psychiatry 2001, 62 (Suppl. S8), 38–45. [Google Scholar] [PubMed]
- Kurina, L.M.; Goldacre, M.J.; Yeates, D.; Gill, L.E. Depression and anxiety in people with inflammatory bowel disease. J. Epidemiol. Community Health 2001, 55, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Cibor, D.; Domagala-Rodacka, R.; Rodacki, T.; Jurczyszyn, A.; Mach, T.; Owczarek, D. Endothelial dysfunction in infammatory bowel diseases: Pathogenesis, assessment and implications. World J. Gastroenterol. 2016, 22, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in infammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar] [CrossRef]
- Tatsuki, M.; Hatori, R.; Nakazawa, T.; Ishige, T.; Hara, T.; Kagimoto, S.; Tomomasa, T.; Arakawa, H.; Takizawa, T. Serological cytokine signature in paediatric patients with infammatory bowel disease impacts diagnosis. Sci. Rep. 2020, 10, 14638. [Google Scholar] [CrossRef]
- Korolkova, O.Y.; Myers, J.N.; Pellom, S.T.; Wang, L.; M’Koma, A.E. Characterization of serum cytokine profile in predominantly colonic inflammatory bowel disease to delineate ulcerative and Crohn’s colitis. Clin. Med. Insights Gastroenterol. 2015, 8, 29–44. [Google Scholar] [CrossRef]
- Shiomi, Y.; Nishiumi, S.; Ooi, M.; Hatano, N.; Shinohara, M.; Yoshie, T.; Kondo, Y.; Furumatsu, K.; Shiomi, H.; Kutsumi, H.; et al. GCMS-based metabolomic study in mice with colitis induced by dextran sulfate sodium. Inflamm. Bowel Dis. 2011, 17, 2261–2274. [Google Scholar] [CrossRef]
- Soares-Mota, M.; Silva, T.A.; Gomes, L.M.; Pinto, M.A.; Mendonça, L.M.; Farias, M.L.; Nunes, T.; Ramalho, A.; Zaltman, C. High prevalence of vitamin A deficiency in Crohn’s disease patients according to serum retinol levels and the relative dose-response test. World J. Gastroenterol. 2015, 21, 1614–1620. [Google Scholar] [CrossRef]
- Parizadeh, S.M.; Jafarzadeh-Esfehani, R.; Hassanian, S.M.; Mottaghi-Moghaddam, A.; Ghazaghi, A.; Ghandehari, M.; Alizade-Noghani, M.; Khazaei, M.; Ghayour-Mobarhan, M.; Ferns, G.A.; et al. Vitamin D in inflammatory bowel disease: From biology to clinical implications. Complement. Ther. Med. 2019, 47, 102189. [Google Scholar] [CrossRef]
- Tahan, G.; Aytac, E.; Aytekin, H.; Gunduz, F.; Dogusoy, G.; Aydin, S.; Tahan, V.; Uzun, H. Vitamin E has a dual effect of anti-inflammatory and antioxidant activities in acetic acid-induced ulcerative colitis in rats. Can. J. Surg. 2011, 54, 333–338. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, R.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Fatty acids from fish: The anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Donnellan, C.F.; Yann, L.H.; Lal, S. Nutritional management of Crohn’s disease. Therap. Adv. Gastroenterol. 2013, 6, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, D.; Rodacki, T.; Domagala-Rodacka, R.; Cibor, D.; Mach, T. Diet and nutritional factors in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ghishan, F.K.; Kiel, P.R. Vitamins and minerals in IBD. Gastroenterol. Clin. N. Am. 2019, 46, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Singleton, C.K.; Martin, P.R. Molecular mechanisms of thiamine utilization. Curr. Mol. Med. 2001, 1, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Dhir, S.; Tarasenko, M.; Napoli, E.; Giulivi, C. Neurological, Psychiatric, and Biochemical Aspects of Thiamine Deficiency in Children and Adults. Front. Psychiatry 2019, 10, 207. [Google Scholar] [CrossRef]
- Butterworth, R.F. Thiamin deficiency and brain disorders. Nutr. Res. Rev. 2003, 16, 277–284. [Google Scholar] [CrossRef]
- Abdou, E.; Hazell, A.S. Thiamine deficiency: An update of pathophysiologic mechanisms and future therapeutic considerations. Neurochem. Res. 2015, 40, 353–361. [Google Scholar] [CrossRef]
- Mikkelsen, K.; Stojanovska, L.; Apostolopoulos, V. The effects of Vitamin B in depression. Curr. Med. Chem. 2016, 23, 4317–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Ding, H.; Chen, H.; Ye, X.; Li, H.; Lin, X.; Ke, Z. Thiamine nutritional status and depressive symptoms are inversely associate among older Chinese adults. J. Nutr. 2013, 143, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Vignisse, J.; Sambon, M.; Gorlova, A.; Pavlov, D.; Caron, N.; Malgrange, B.; Shevtsova, E.; Svistunov, A.; Anthony, D.C.; Markova, N.; et al. Thiamine and benfotiamine prevent stress-induced suppression of hippocampal neurogenesis in mice exposed to predation without affecting brain thiamine diphosphate levels. Mol. Cell Neurosci. 2017, 82, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Kishimoto, T.; Oishi, S.; Nagata, K.; Hasegawa, S.; Watanabe, T.; Kida, S. Vitamin B1-deficient mice show impairment of hippocampus-dependent memory formation and loss of hippocampal neurons and dendritic spines: Potential microendophenotypes of Wernicke-Korsakoff syndrome. Biosci. Biotechnol. Biochem. 2016, 80, 2425–2436. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann. Neurol. 1992, 31, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; De Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Choi, D.W. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988, 11, 465–469. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Teng, T.; Jiang, Y.; Xiang, Y.; Fan, L.; Yu, Y.; Zhou, X.; Xie, P. Comparative analysis of gut microbiota and fecal metabolome features among multiple depressive animal models. J. Affect. Disord. 2022, 314, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.-H.; Zhao, Y.-Q.; Wu, H.T.; Fan, M.; Zhu, L.L. Cortical inflammation is increased in a DSS-induced colitis mouse model. Neurosci. Bull. 2018, 34, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Haj-Mirzaian, A.; Amiri, S.; Amini-Khoei, H.; Hosseini, M.-J.; Haj-Mirzaian, A.; Momeny, M.; Rahimi-Balaei, M.; Dehpour, A.R. Anxiety- and depressive-like behaviors are associated with altered hippocampal energy and infammatory status in a mouse model of Crohn’s disease. Neuroscience 2017, 366, 124–137. [Google Scholar] [CrossRef]
- Green, H.F.; Treacy, E.; Keohane, A.K.; Sullivan, A.M.; O’Keeffe, G.W.; Nolan, Y.M. A role for interleukin-1β in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol. Cell Neurosci. 2012, 49, 311–321. [Google Scholar] [CrossRef]
- Boehme, M.; Guenther, M.; Stahr, A.; Liebmann, M.; Jaenisch, N.; Witte, O.W.; Frahm, C. Impact of indomethacin on neuroinflammation and hippocampal neurogenesis in aged mice. Neurosci. Lett. 2014, 572, 7–12. [Google Scholar] [CrossRef]
- Vallières, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Hashimoto, K. Inflammatory biomarkers 1 as differential predictors of antidepressant response. Int. J. Mol. Sci. 2015, 16, 7796–7801. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res. Rev. 2009, 61, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with C-reactive protein IL-1 and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Na, K.S.; Shin, K.H.; Jung, H.Y.; Choi, S.H.; Kim, J.B. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1044–1053. [Google Scholar] [CrossRef]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.-C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.; Miller, H.L.; Charney, D.S. Hippocampal Volume Reduction in Major Depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef]
- Chen, J.Y.; Blankstein, U.; Diamant, N.E.; Davis, K.D. White matter abnormalities in irritable bowel syndrome and relation to individual factors. Brain Res. 2011, 1392, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Niddam, D.M.; Tsai, S.-Y.; Lu, C.-L.; Ko, C.-W.; Hsieh, J.-C. Reduced Hippocampal Glutamate–Glutamine Levels in Irritable Bowel Syndrome: Preliminary Findings Using Magnetic Resonance Spectroscopy. Am. J. Gastroenterol. 2011, 106, 1503–1511. [Google Scholar] [CrossRef]
- Mehrian-Shai, R.; Reichardt, J.; Harris, C.C.; Toren, A. The Gut–Brain Axis, Paving the Way to Brain Cancer. Trends Cancer 2019, 5, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Braganhol, E.; Kukulski, F.; Lévesque, S.A.; Fausther, M.; Lavoie, E.G.; Zanotto-Filho, A.; Bergamin, L.S.; Pelletier, J.; Bahrami, F.; Ben Yebdri, F.; et al. Nucleotide receptors control IL-8/CXCL8 and MCP-1/CCL2 secretions as well as proliferation in human glioma cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 120–130. [Google Scholar] [CrossRef] [PubMed]
- McFarland, B.C.; Hong, S.W.; Rajbhandari, R.; Twitty, G.B., Jr.; Gray, G.K.; Yu, H.; Benveniste, E.N.; Nozell, S.E. NF-κB-Induced IL-6 Ensures STAT3 Activation and Tumor Aggressiveness in Glioblastoma. PLoS ONE 2013, 8, e78728. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Béliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 318, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; Woo, J. From Gut to Brain: Alteration in Inflammation Markers in the Brain of Dextran Sodium Sulfate-induced Colitis Model Mice. Clin. Psychopharmacol. Neurosci. 2018, 16, 422–433. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Tanori, M.; Mancuso, M.; Gessi, M.; Pasquali, E.; Leonardi, S.; Oliva, M.A.; Rebessi, S.; Di Majo, V.; Covelli, V.; et al. Two-hit model for progression of medulloblastoma preneoplasia in Patched heterozygous mice. Oncogene 2006, 25, 5575–5580. [Google Scholar] [CrossRef]
- Selvadurai, H.J.; Luis, E.; Desai, K.; Lan, X.; Vladoiu, M.C.; Whitley, O.; Galvin, C.; Vanner, R.J.; Lee, L.; Whetstone, H.; et al. Medulloblastoma Arises from the Persistence of a Rare and Transient Sox2+ Granule Neuron Precursor. Cell Rep. 2020, 31, 107511. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Walton, K.L.W.; Blue, E.; McNaughton, K.K.; Magness, S.T.; Lund, P.K. Mucosal Healing and Fibrosis after Acute or Chronic Inflammation in Wild Type FVB-N Mice and C57BL6 Procollagen α1(I)-Promoter-GFP Reporter Mice. PLoS ONE 2012, 7, e42568. [Google Scholar] [CrossRef]
- Coppola, M.; Diretto, G.; Digilio, M.C.; Woo, S.L.; Giuliano, G.; Molisso, D.; Pennacchio, F.; Lorito, M.; Rao, R. Transcriptome and Metabolome Reprogramming in Tomato Plants by Trichoderma harzianum strain T22 Primes and Enhances Defense Responses Against Aphids. Front. Physiol. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, M.; Mumm, R.; Vos, R.C.H.; Ruan, J. Metabolomics reveals the within-plant spatial effects of shading on tea plants. Tree Physiol. 2020, 41, 317–330. [Google Scholar] [CrossRef] [PubMed]
- García-Domínguez, X.; Diretto, G.; Peñaranda, D.S.; Frusciante, S.; García-Carpintero, V.; Cañizares, J.; Vicente, J.S.; Marco-Jiménez, F. Early Embryo Exposure to Assisted Reproductive Manipulation Induced Subtle Changes in Liver Epigenetics with No Apparent Negative Health Consequences in Rabbit. Int. J. Mol. Sci. 2021, 22, 9716. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001, 4, 1–9. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Metabolite | Formula | Adduct | FC DSS/UN | ||
|---|---|---|---|---|---|---|
| AC-7 | AC-14 | CC | ||||
| Amines | N-acetyloctopamine | C10H13NO3 | [M-H]−1 | −0.11 | −0.27 * | −0.27 * |
| Amino Acids, Peptides and Proteins | Thyronine | C15H15NO4 | [M+H]+1 | −1.08 | −1.50 | −1.16 * |
| L-SerineO-sulfate | C3H7NO6S | [M+FA-H]− | −1.84 * | −0.95 | −0.42 | |
| Val-Val | C10H20 N2O3 | [M+H]+1 | −1.13 * | −0.03 | 0.81 | |
| 3,5-Diiodo-L-tyrosine | C8H9 I2 NO | [M+H]+1 | −1.46 * | −0.71 | −0.77 | |
| N-lauroyl glycine | C14H27 NO3 | [M+H]+1 | −0.86 | −1.02 | −1.69 * | |
| D-Aspartic acid | C4H7NO4 | [M+H]+1 | −0.42 | −0.04 | −0.87 * | |
| Fatty Acyls [FA] | (S)-N-(1-amino-1-oxo-3-phenylpropan-2-yl)oleamide | C27H45NO3 | [M+H-H2O]+ | −0.95 | −1.62 | −2.10 * |
| methyl 8-[2-(2-formyl-vinyl)-3-hydroxy-5-oxo-cyclopentyl]-octanoate | C17H26O5 | [M+H]+1 | −0.29 | −0.48 * | −0.37 * | |
| Pimelic acid | C7H12O4 | [M-H]−1 | −0.68 | −0.61 | −1.79 * | |
| Suberic acid | C8H14O4 | [M-H]−1 | −0.47 | −0.32 | −2.08 * | |
| Azelaic acid | C9H16O4 | [M-H]−1 | −0.27 | −0.19 | −1.79 * | |
| Oleoyl Ethanolamide | C20H39NO2 | [M+H]+1 | −2.14 * | −1.32 | −1.48 * | |
| Linoleyl carnitine | C25H45 NO4 | [M+H]+1 | −2.73 * | −1.93 | 0.13 | |
| Dihydrozeatin-O-glucoside-like | C16H25 N5O6 | [M-H]−1 | −2.19 | 0.81 | −1.31 * | |
| 15-Deoxy-Δ12.14-prostaglandin J2-2-glycerol ester | C23H34O5 | [M+H]+1 | −0.63 | −0.98 | −2.18 * | |
| 9Z-Eicosenoic acid | C14H30O8 | [M+NH4]+1 | −0.68 | −1.06 | −0.23 * | |
| Hexadecanedioic acid Mono-L-carnitine ester | C23H43 NO6 | [M+H]+1 | −0.81 | −0.91 | −2.17 * | |
| Palmitoleoyl-EA | C18H35 NO2 | [M+H]+1 | −1.42 | −1.28 | −1.62 * | |
| Oleamide | C18H35 NO | [M+H]+1 | −1.33 | −1.64 | −1.15 * | |
| Octadecanamide | C18H37 NO | [M+H]+1 | −1.42 | −1.50 * | −1.19 * | |
| Glycerophospholipids | LysoPC(18:1(9Z)) | C26H52NO7 P | [M+H]+1 | 0.16 | −0.27 | −1.13 * |
| Phospholipids | PI(O-16:0/0:0) | C32H44O7 | [M-H]−1 | −0.35 | −2.13 | −1.60 * |
| Prenol Lipids | 3.4-didehydro-retinoate | C20H26O2 | [M-H]−1 | −0.05 | 0.05 | −0.37 * |
| 9-cis-Retinoic acid | C20H28O2 | [M-H]−1 | −0.47 | −0.12 | −2.00 * | |
| Alpha-Tocotrienol | C29H44O2 | [M+H]+1 | −0.86 | −1.19 | −1.75 * | |
| Ecabet-like | C20H28O5S | [M-H]−1 | −3.21 * | −1.82 | −1.34 * | |
| Sterol lipids | ST 28:2;O7 | C28H46O7 | [M+Na]+ | −1.47 | −2.05 * | −2.41 * |
| Abrusoside A-like | C36H54O10 | [M-H]−1 | −1.67 * | −1.12 | −0.67 | |
| 1-a.24R.25-Trihydroxyvitamin D2 | C28H44O4 | [M+CH3COO]− | 0.74 | 1.45 | −0.89 | |
| 1α.25-dihydroxy-26.27-dimethyl-20.21-methano-23-oxavitamin D3/1α.25-dihydroxy-26.27-dimethyl-20.21-methano-23-oxacholecalciferol | C29H46O4 | [M+FA-H]− | −1.76 | −1.61 | −1.93 * | |
| 11b-hydroxyprogesterone | C20H28O4 | [M-H]−1 | −2.17 | −0.36 | −1.95 * | |
| 16alpha.17-Isopropylidenedioxy-6alPha-methylprogesterone | C25H36O4 | [M+CH3COO]− | −0.60 * | −0.58 | −0.89 * | |
| Sterol lipids/Bile acid | Lithocholic acid glycine conjugate | C26H43 NO4 | [M+H]+1 | −0.88 | −1.14 | −1.61 * |
| Phenylpropanoids and polyketides | Afrormosin-like | C17H14O5 | [M+H]+1 | −0.56 | −0.95 | −1.51 * |
| Bergaptol-like | C11H6O4 | [M+H-H2O]+1 | −1.04 * | −1.16 * | −0.44 | |
| Organoheterocyclic compounds | 5-(2-Hydroxyethyl)-4-methylthiazole | C6H9NOS | [M+H]+1 | −0.90 | −0.71 | −1.13 * |
| Thiamine | C12H16N4OS | [M+2H]+2 | −0.70 | −0.32 | −1.03 * | |
| Iprodione-like | C13H13Cl2N3O3 | [M-H]−1 | −0.00 | 0.28 | −1.27 * | |
| Mesobilirubinogen | C33H44N4O6 | [M-H]−1 | −0.64 | −1.57 | −1.49 * | |
| Organic Nitrogen compounds | N,N-Dimethylsphing-4-enine | C20H41NO2 | [M+H]+1 | −1.20 | −2.32 * | −1.18 * |
| Organic acids and derivative | Tazobactam-like | C10H12N4O5S | [M+H-H2O]+1 | 0.07 | 0.26 | −1.21 * |
| 4-Hydroxyphenylacetylglycine | C10H11NO4 | [M-H]−1 | 1.66 * | 1.57 | 0.66 | |
| Nucleosides, Nucleotides and analogues | 5’-Deoxyadenosine | C10H13N5O3 | [M+H]+1 | −1.12 * | −1.55 * | −0.89 |
| 4-Amino-5-hydroxymethyl-2-methylpyrimidine | C6H9 N3O | [M+H]+1 | −1.45 | −1.13 | −1.03 * | |
| Benzenoids | Phenylethylamine | C14H27 NSi2 | [M+H]+1 | −1.06 | −1.34 | −1.45 * |
| Metoclopramide-like | C14H22ClN3O2 | [M+H]+1 | −0.94 | −1.03 | −2.07 * | |
| Butylparaben-like | C11H14O3 | [M-H]−1 | −1.24 | −0.82 | −1.50 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitali, R.; Prioreschi, C.; Lorenzo Rebenaque, L.; Colantoni, E.; Giovannini, D.; Frusciante, S.; Diretto, G.; Marco-Jiménez, F.; Mancuso, M.; Casciati, A.; et al. Gut–Brain Axis: Insights from Hippocampal Neurogenesis and Brain Tumor Development in a Mouse Model of Experimental Colitis Induced by Dextran Sodium Sulfate. Int. J. Mol. Sci. 2022, 23, 11495. https://doi.org/10.3390/ijms231911495
Vitali R, Prioreschi C, Lorenzo Rebenaque L, Colantoni E, Giovannini D, Frusciante S, Diretto G, Marco-Jiménez F, Mancuso M, Casciati A, et al. Gut–Brain Axis: Insights from Hippocampal Neurogenesis and Brain Tumor Development in a Mouse Model of Experimental Colitis Induced by Dextran Sodium Sulfate. International Journal of Molecular Sciences. 2022; 23(19):11495. https://doi.org/10.3390/ijms231911495
Chicago/Turabian StyleVitali, Roberta, Clara Prioreschi, Laura Lorenzo Rebenaque, Eleonora Colantoni, Daniela Giovannini, Sarah Frusciante, Gianfranco Diretto, Francisco Marco-Jiménez, Mariateresa Mancuso, Arianna Casciati, and et al. 2022. "Gut–Brain Axis: Insights from Hippocampal Neurogenesis and Brain Tumor Development in a Mouse Model of Experimental Colitis Induced by Dextran Sodium Sulfate" International Journal of Molecular Sciences 23, no. 19: 11495. https://doi.org/10.3390/ijms231911495
APA StyleVitali, R., Prioreschi, C., Lorenzo Rebenaque, L., Colantoni, E., Giovannini, D., Frusciante, S., Diretto, G., Marco-Jiménez, F., Mancuso, M., Casciati, A., & Pazzaglia, S. (2022). Gut–Brain Axis: Insights from Hippocampal Neurogenesis and Brain Tumor Development in a Mouse Model of Experimental Colitis Induced by Dextran Sodium Sulfate. International Journal of Molecular Sciences, 23(19), 11495. https://doi.org/10.3390/ijms231911495