
Transcriptomic Analysis to Unravel Potential Pathways and Genes Involved in Pecan (Carya illinoinensis) Resistance to Pestalotiopsis microspora

Abstract
:1. Introduction
2. Results
2.1. Evaluation of Pecan Fruits against FBS Infection
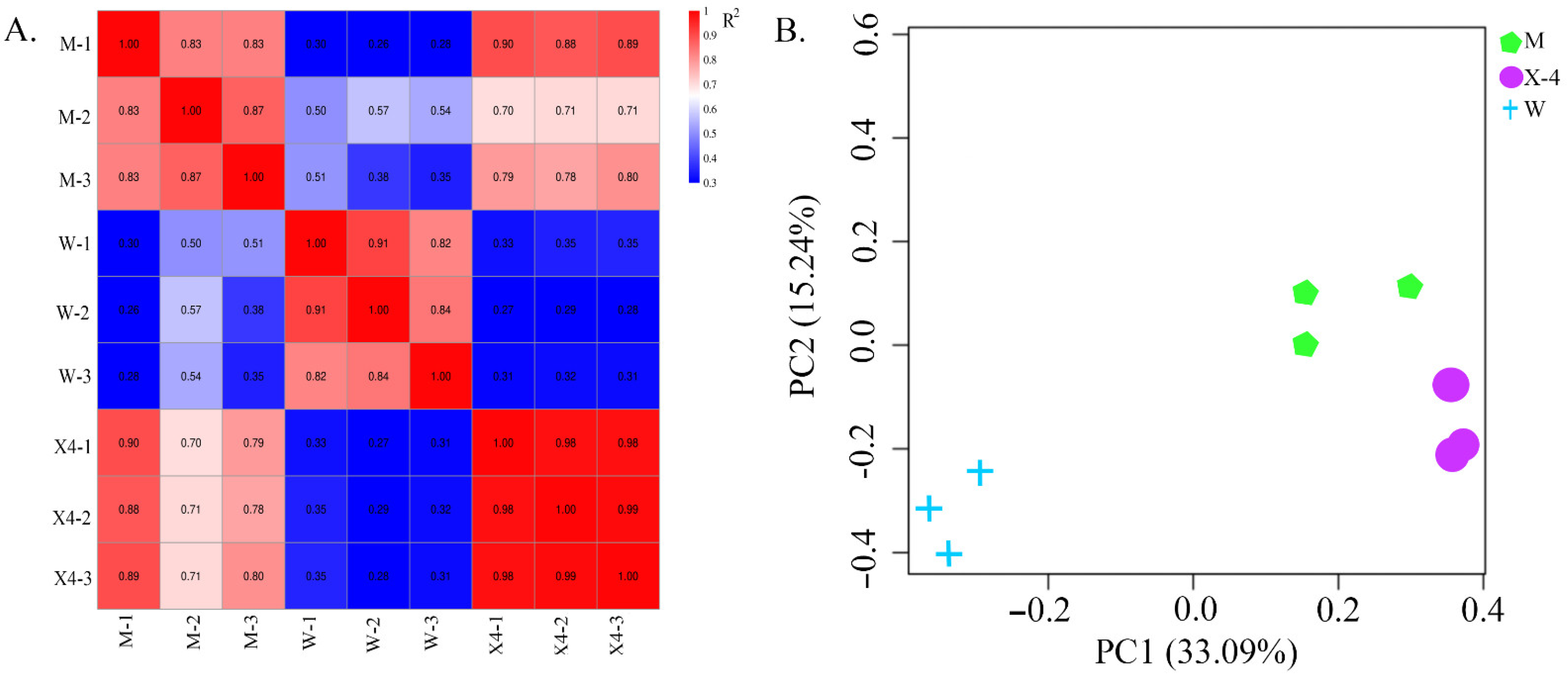
2.2. Library Construction and Sequencing
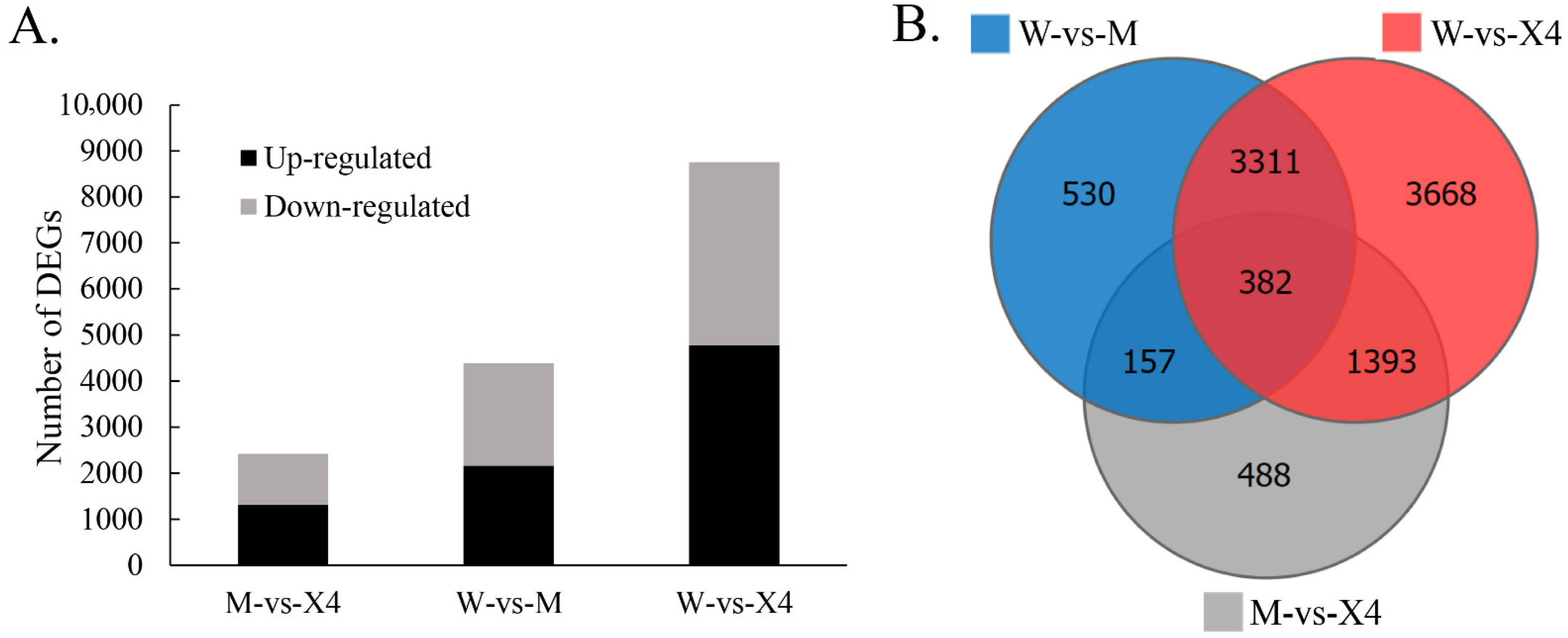
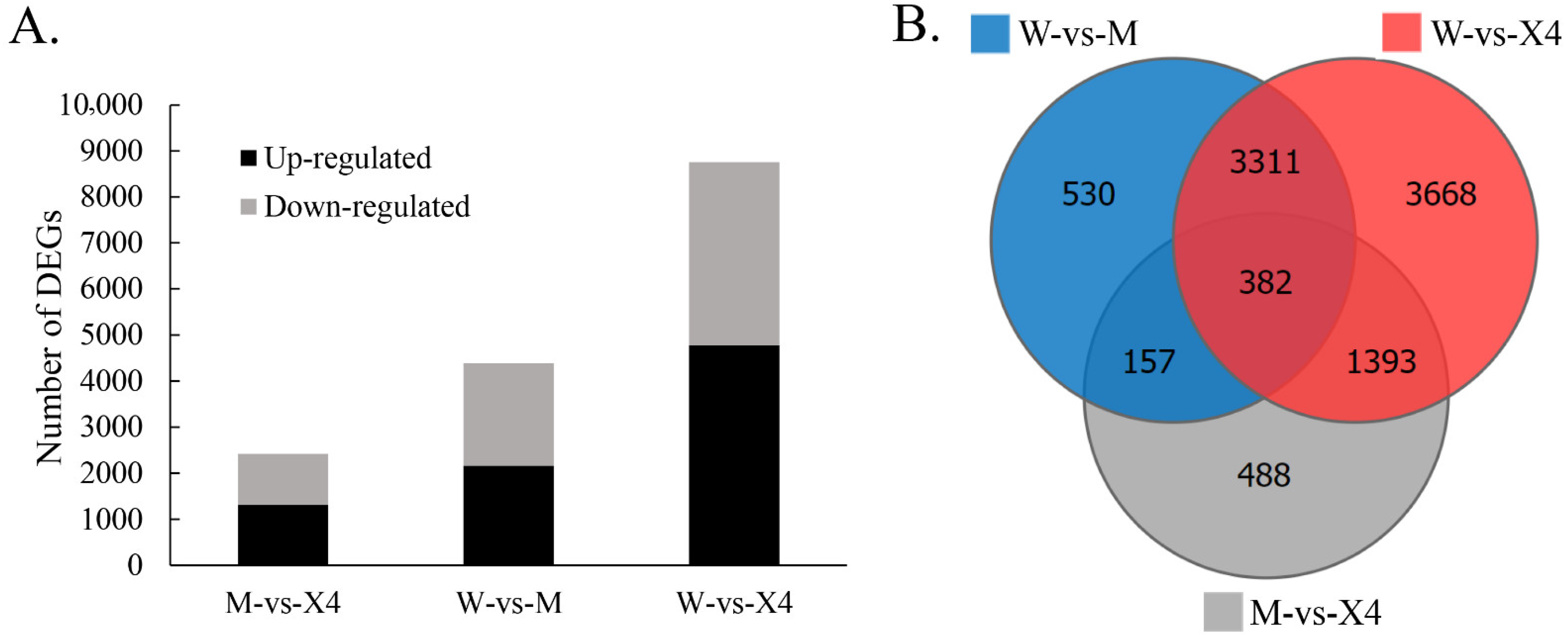
2.3. Differentially Expressed Genes (DEGs)
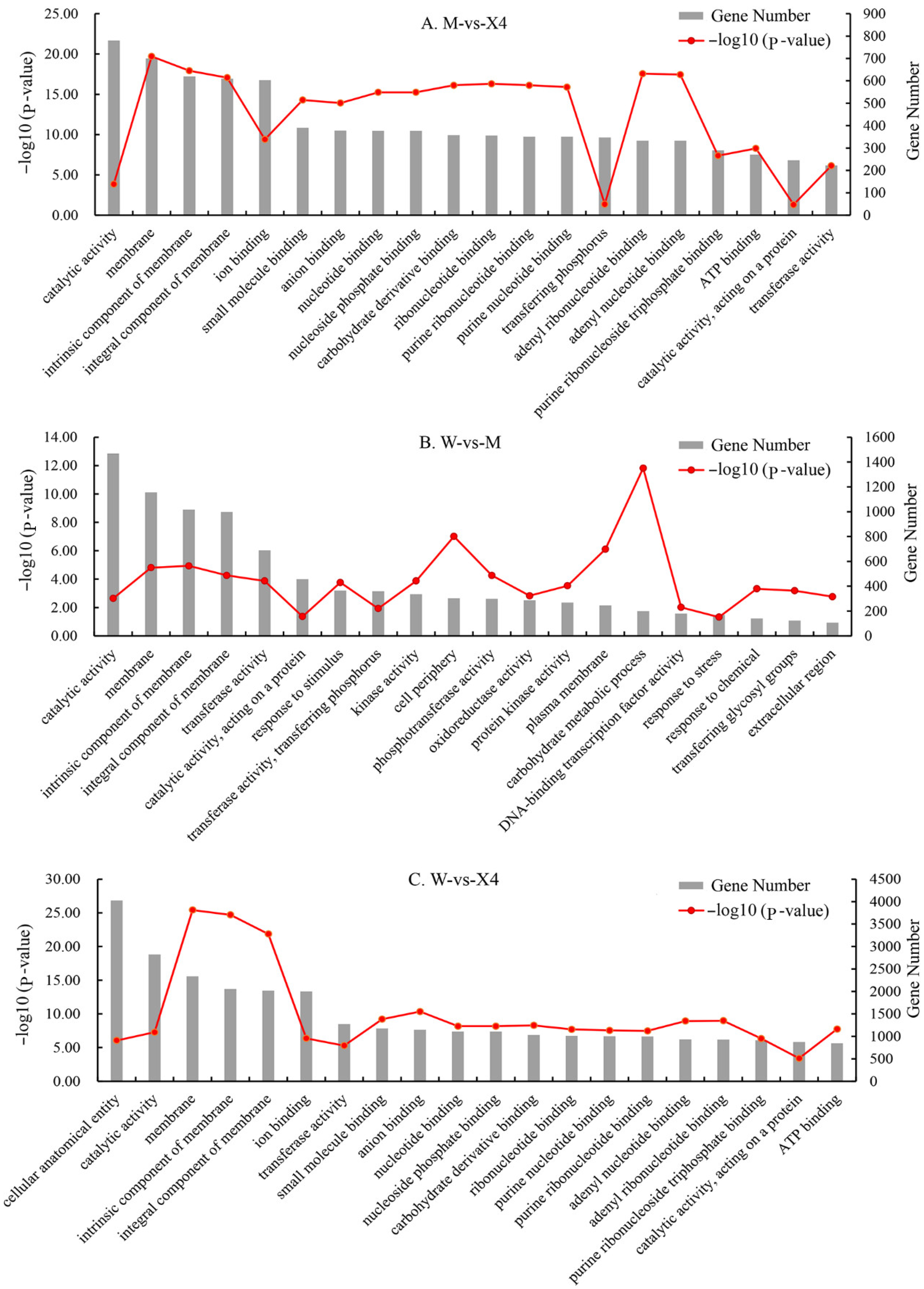
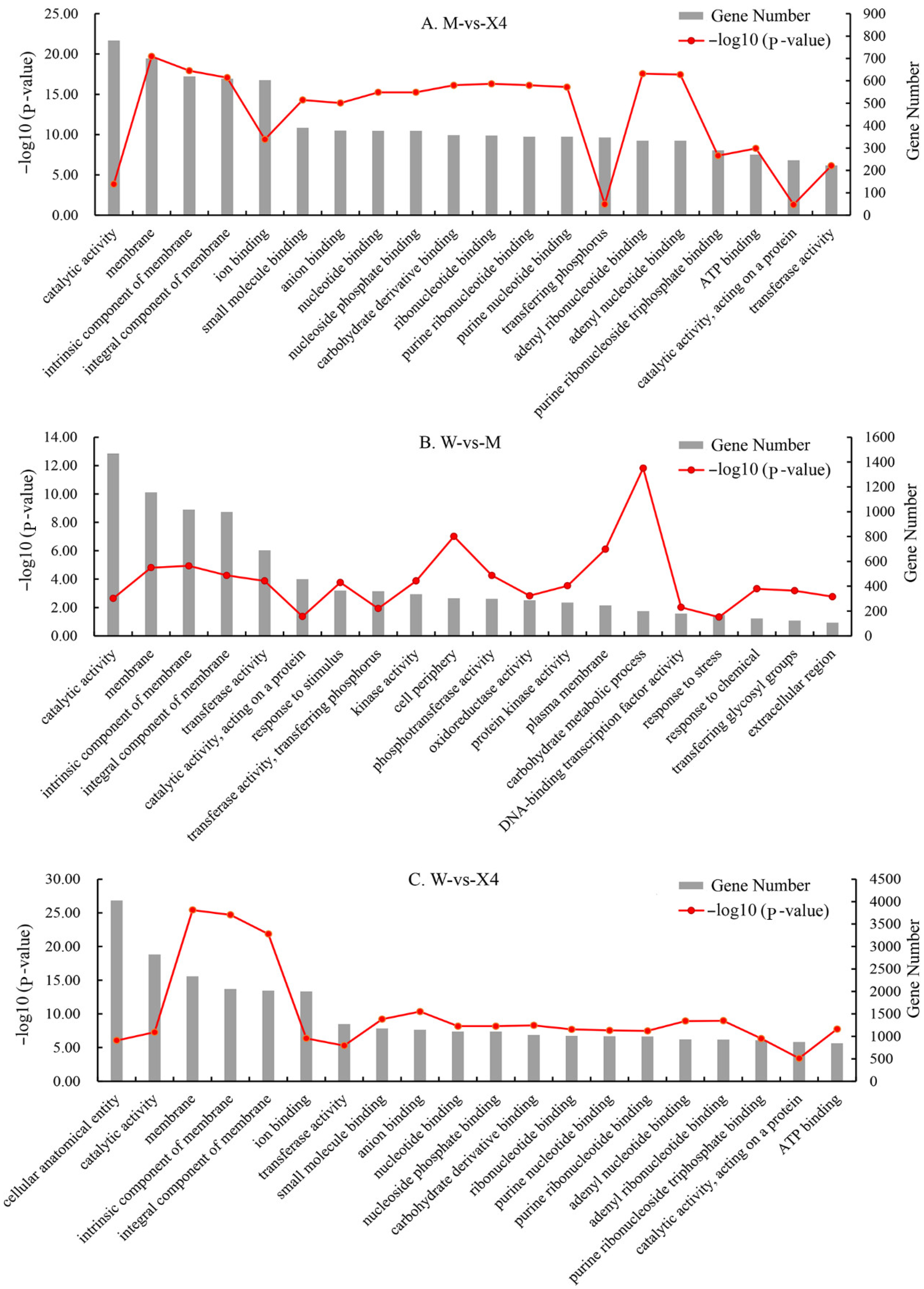
2.4. GO and KEGG Functional Enrichment Analysis of DEGs
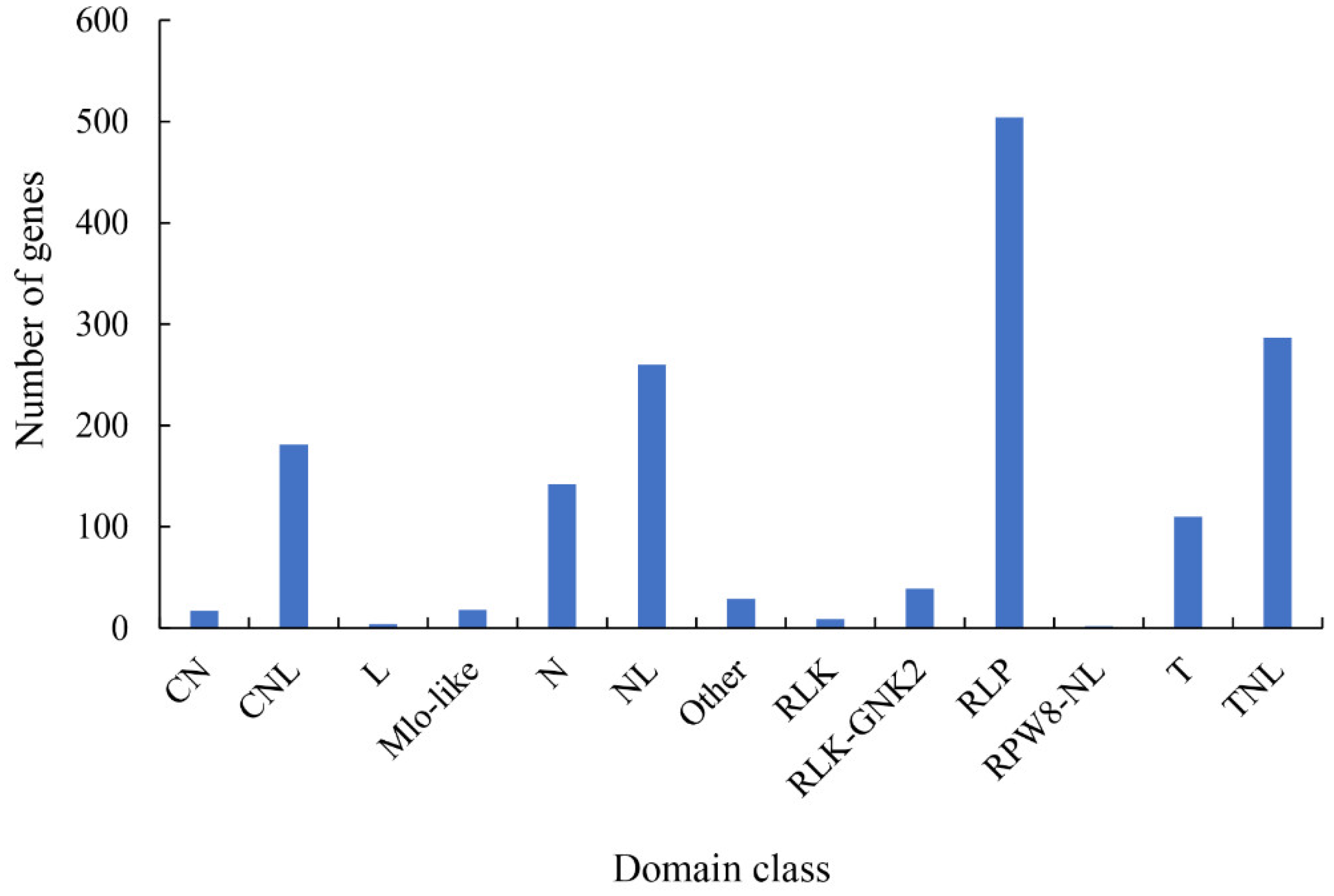
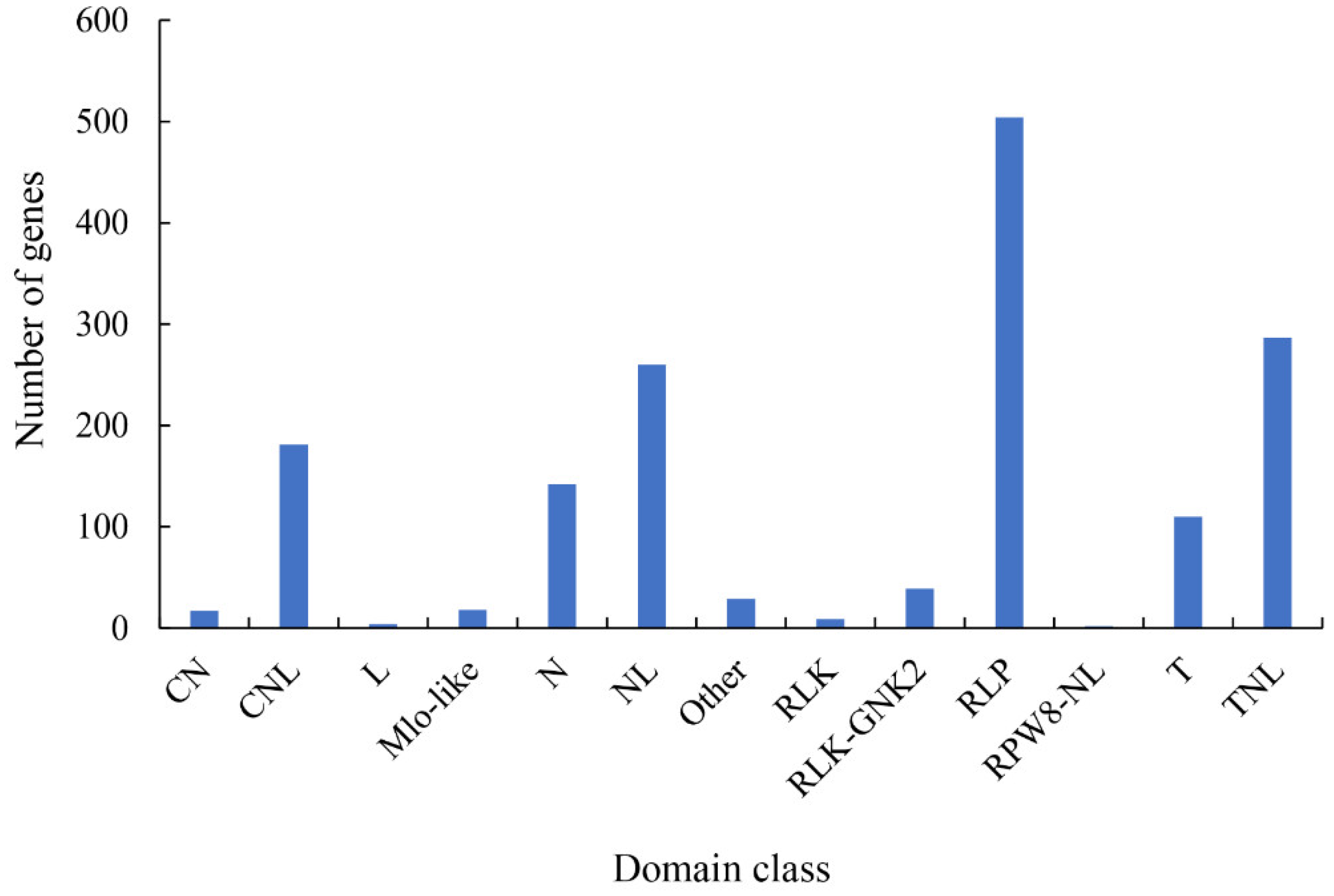
2.5. Pathogen-Related Genes (PRGs)
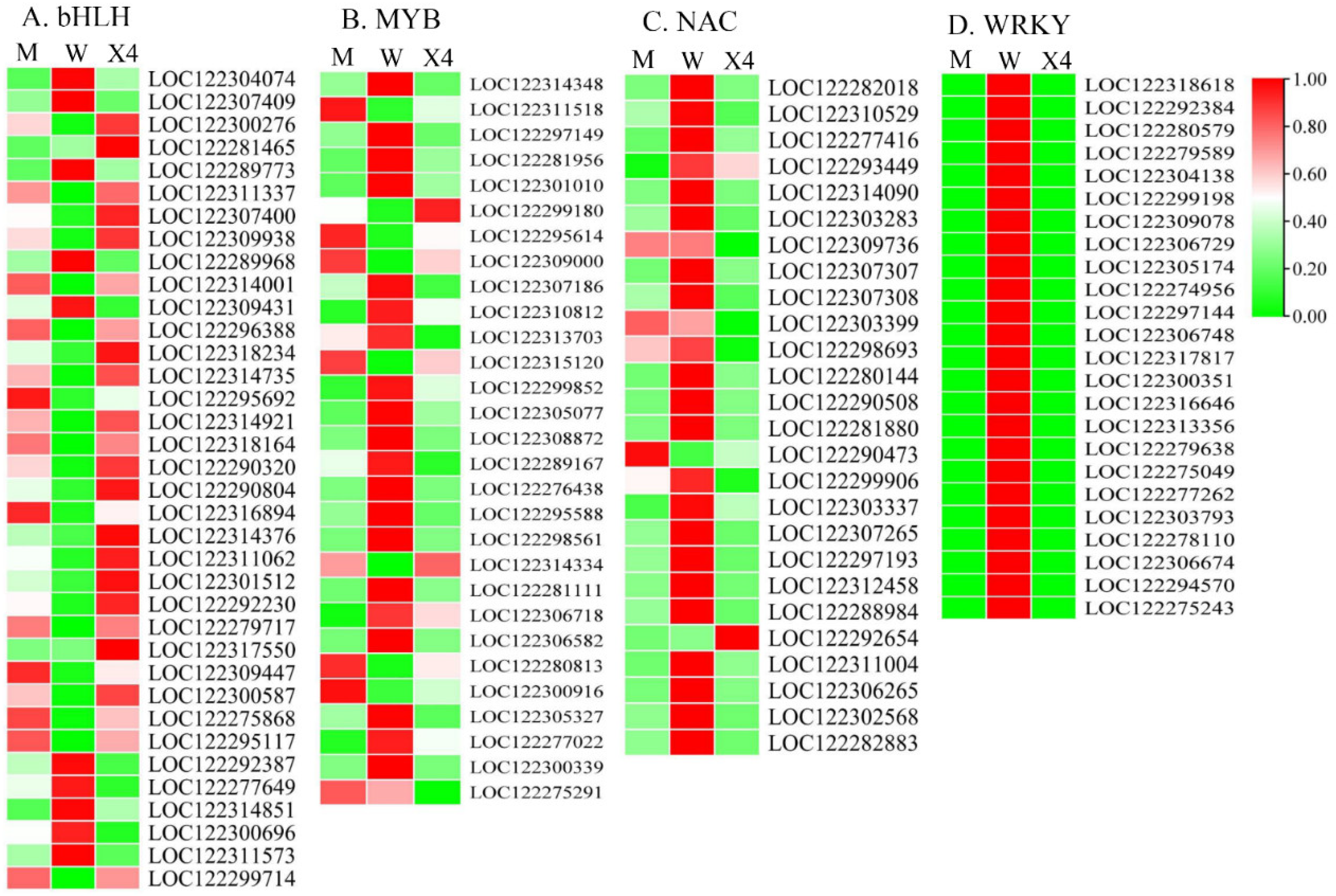
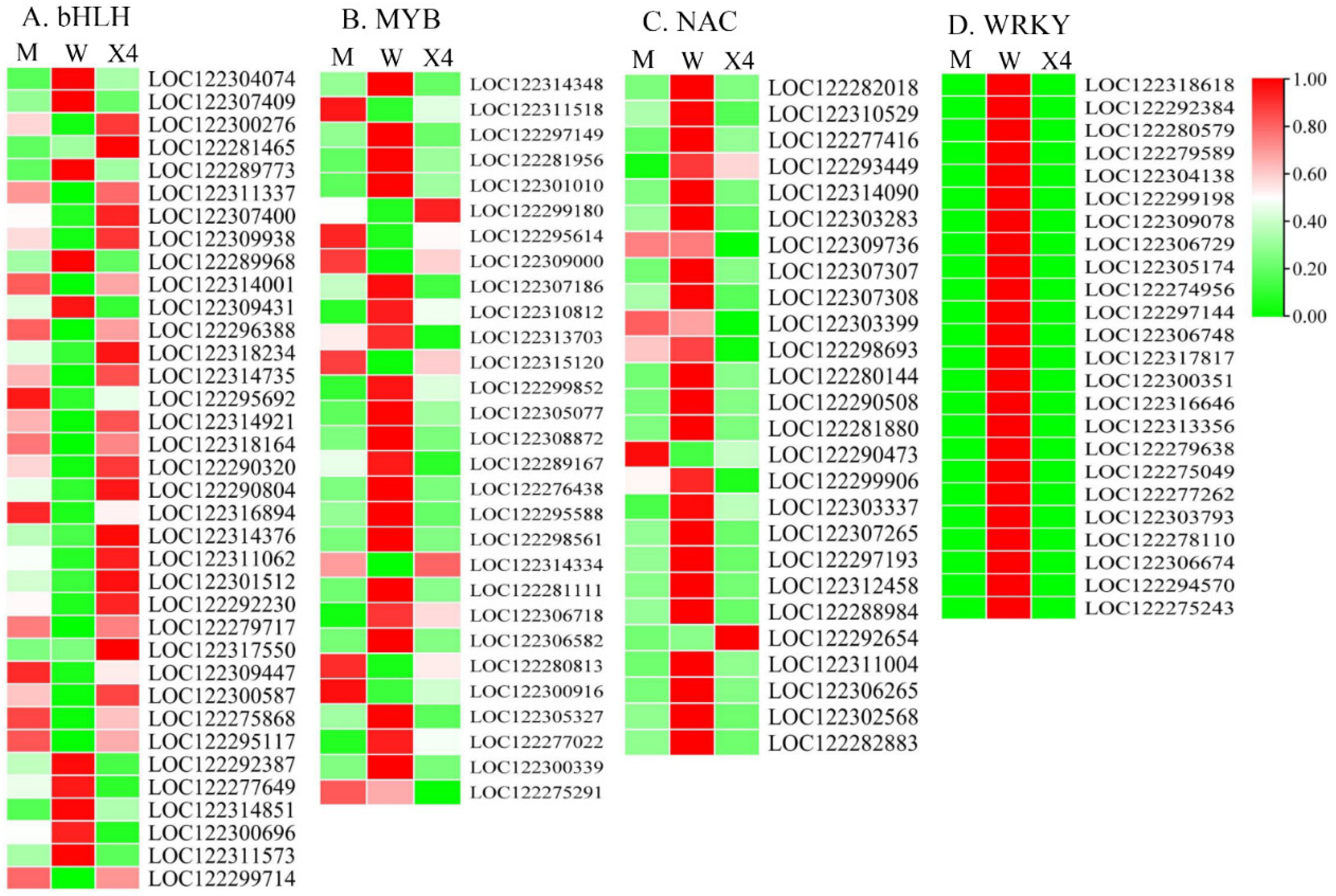
2.6. Transcription Factor Analysis
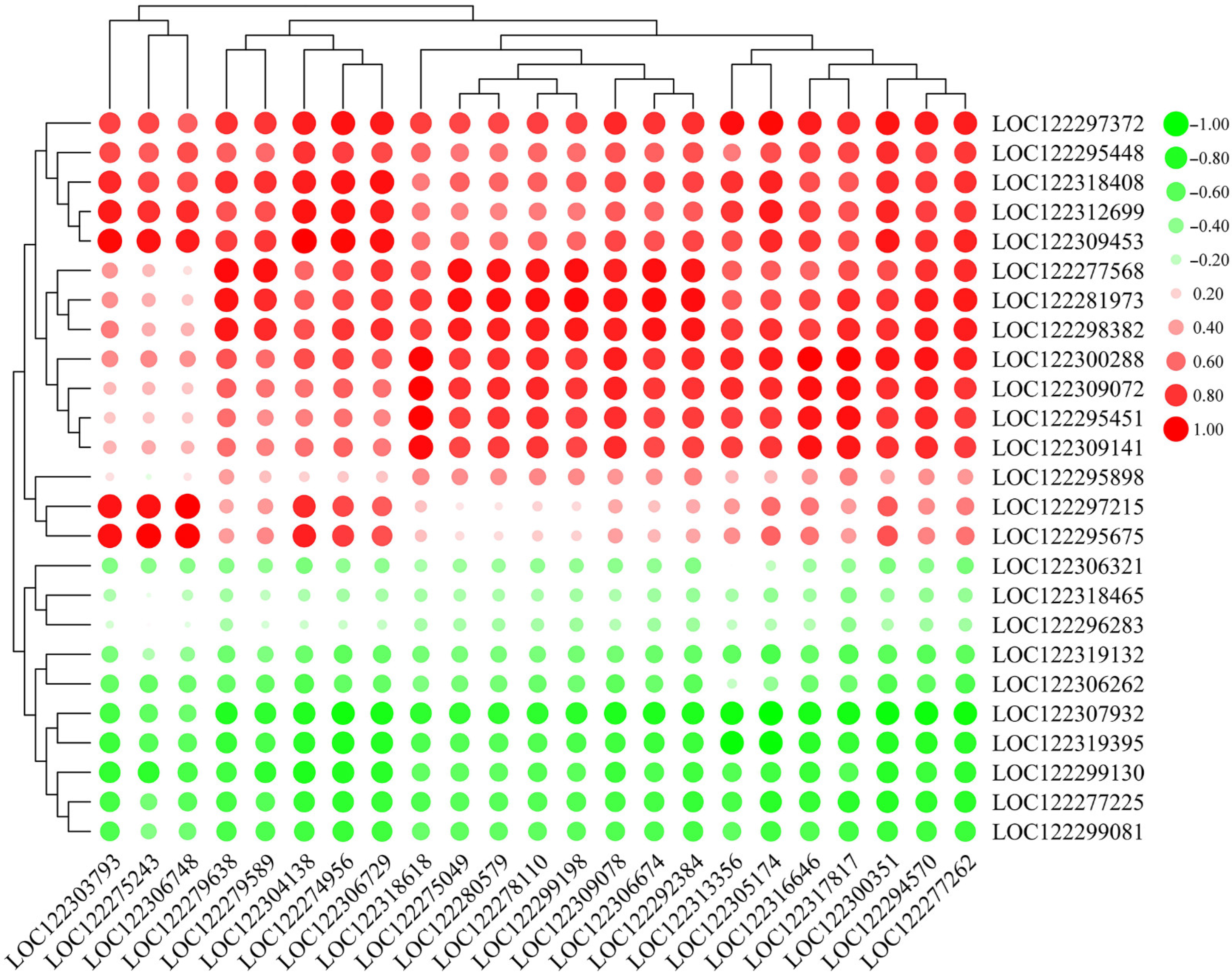
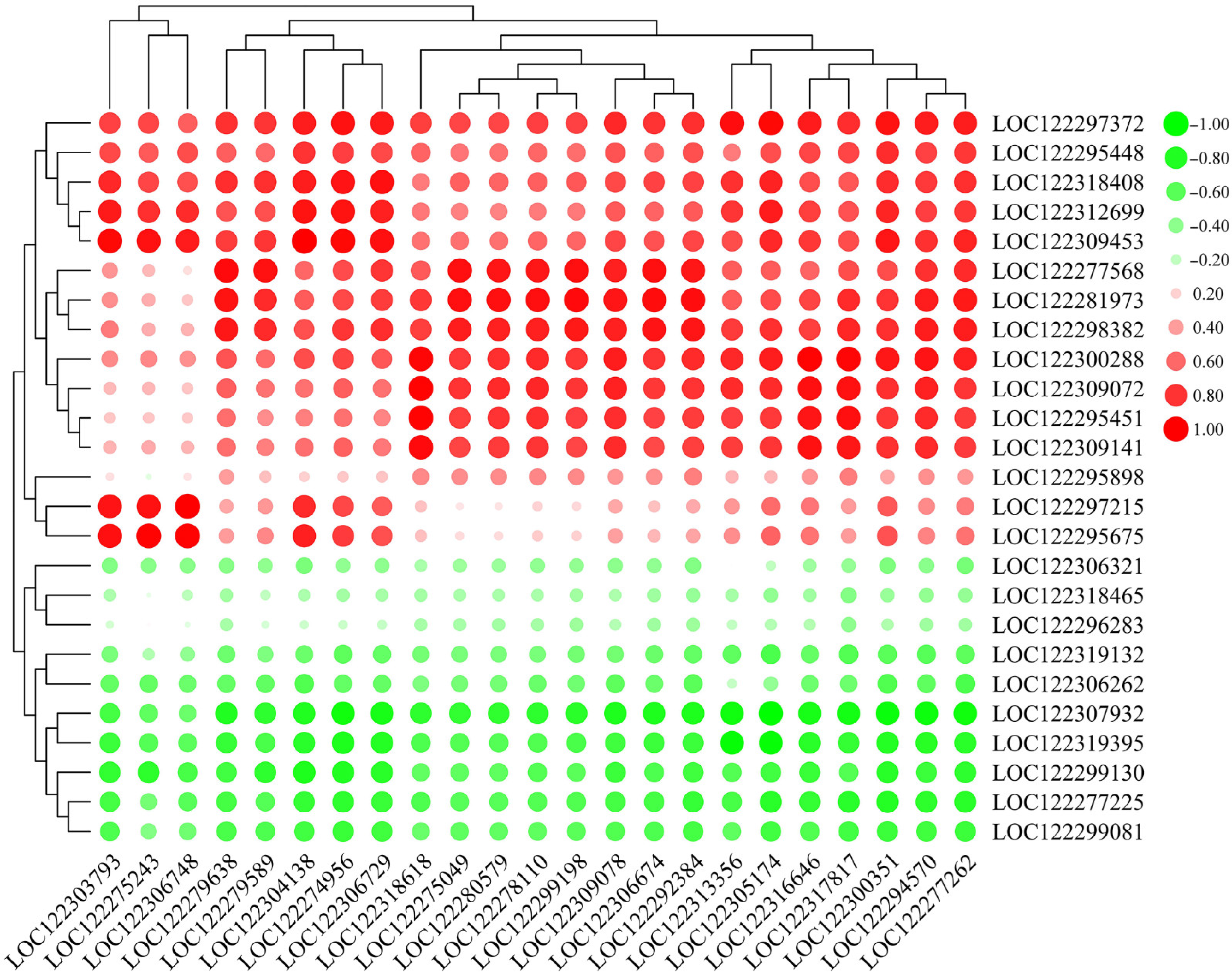
2.7. Correlation of Pathogen-Related Genes (PRGs) and WRKY TFs
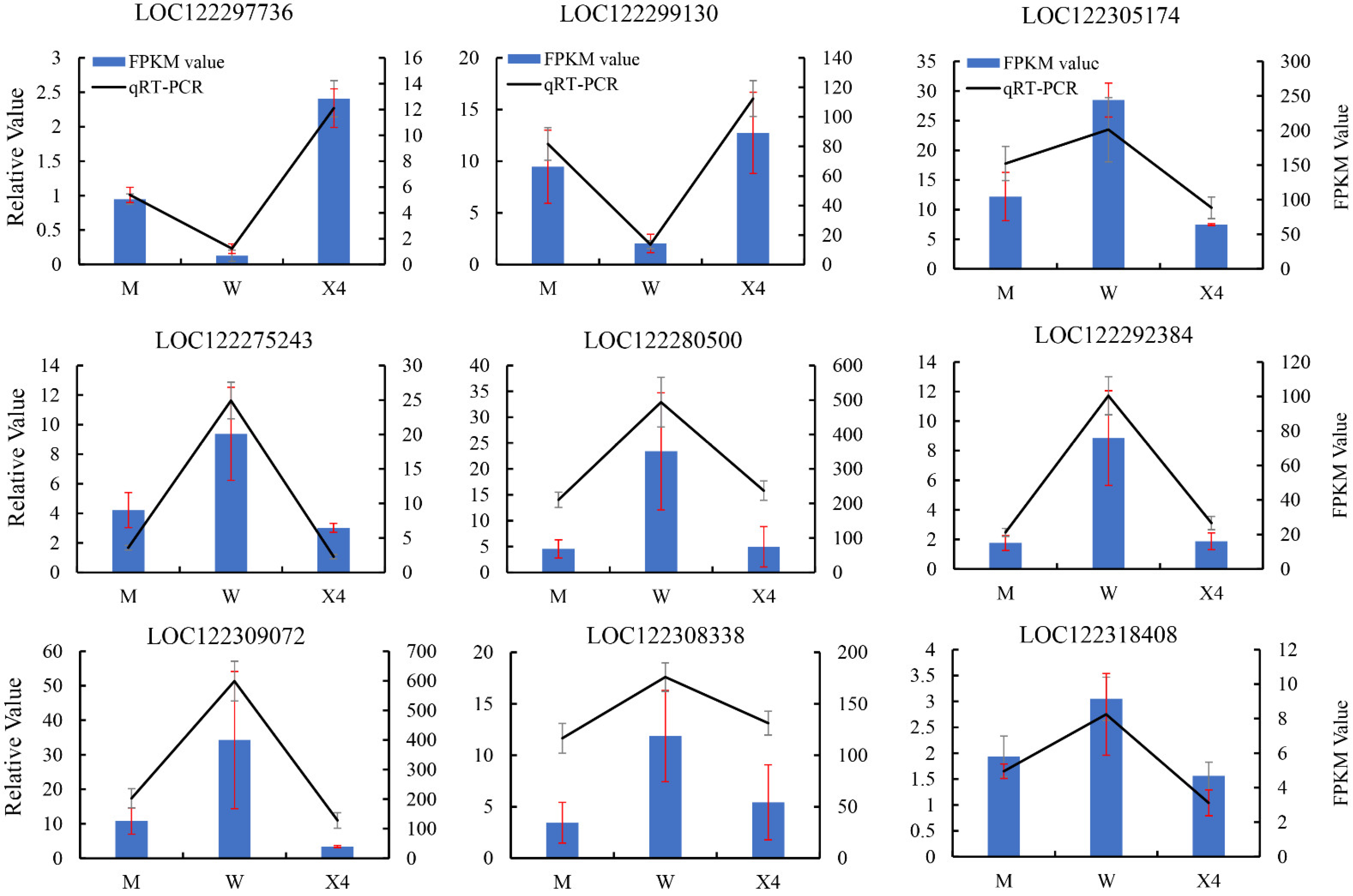
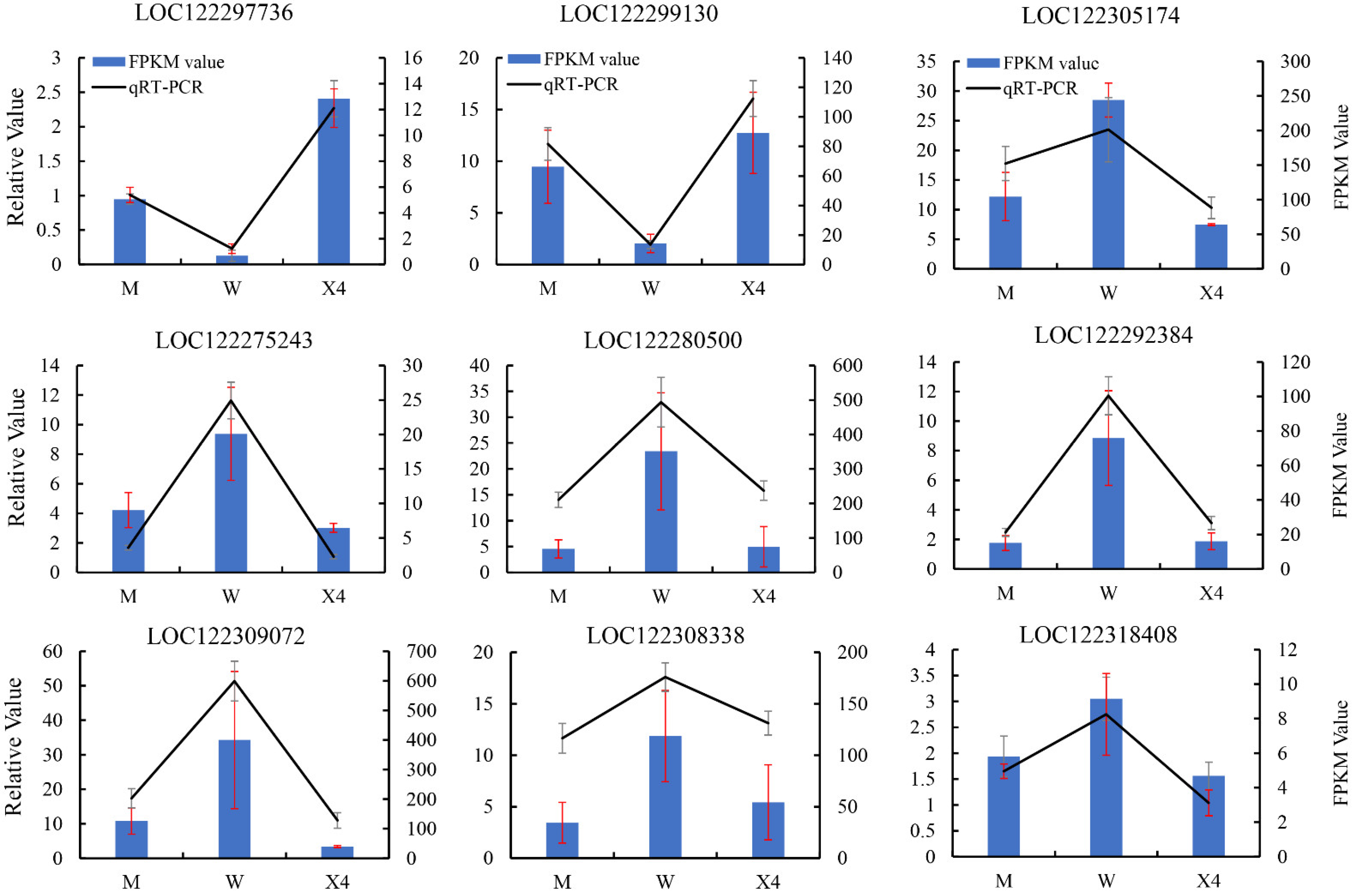
2.8. Confirmation of DEGs by qRT-PCR
3. Discussion
4. Material and Methods
4.1. Plant Material and Sample Collection
4.2. RNA Preparation and Transcriptome Sequencing
4.3. RNA-Seq Data and Differentially Expressed Gene (DEG) Analysis
4.4. GO and KEGG Enrichment Annotation of DEGs
4.5. Screening of Transcription Factors (TFs)
4.6. Identification of Pathogen-Related Genes (PRGs)
4.7. Correlation Network Analysis of PRGs and WRKY TFs
4.8. Candidate DEG Validation by qRT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mo, Z.; Feng, G.; Su, W.; Liu, Z.; Peng, F. Transcriptomic analysis provides insights into grafting union development in pecan (Carya illinoinensis). Genes 2018, 9, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Zhang, C.; Shan, L.; Xu, K.; Xu, J.; Qi, Q.; Xu, Z. First report of Pestalotiopsis microspora as a causal agent of black spot of pecan (Carya illinoinensis) in China. Plant Dis. 2015, 99, 1276. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, J.; Pan, C. Integrated physiological, proteomic, and metabolomic analyzes of pecan cultivar ‘Pawnee’ adaptation to salt stress. Sci. Rep. 2022, 12, 1841. [Google Scholar] [CrossRef]
- Zhang, J.; Jiao, Y.; Sharma, A.; Shen, D.; Wei, B.; Hong, C.; Zheng, B.; Pan, C. Transcriptomic analysis reveals potential pathways associated with salt resistance in pecan (Carya illinoensis K. Koch). J. Biotechnol. 2021, 330, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yao, X.; Zeng, Y.; Zhang, C. Genome-wide identification, characterization, and expression profiling of AP2/ERF superfamily genes under different development and abiotic stress conditions in pecan (Carya illinoinensis). Int. J. Mol. Sci. 2022, 23, 2920. [Google Scholar] [CrossRef]
- Sanogo, S.; Lujan, P. Rarity of a fungal pathogen and a parasitic flowering plant versus the commonness of a mycorrhizal fungus in pecan orchards in New Mexico. Plant Health Prog. 2018, 19, 207–211. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.-A. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infect. Immun. 1999, 67, 3703–3713. [Google Scholar] [CrossRef] [Green Version]
- Navarre, W.W.; Zychlinsky, A. Pathogen-induced apoptosis of macrophages: A common end for different pathogenic strategies: Microreview. Cell. Microbiol. 2000, 2, 265–273. [Google Scholar] [CrossRef]
- Deng, B.; Wang, W.; Deng, L.; Yao, S.; Ming, J.; Zeng, K. Comparative RNA-Seq analysis of citrus fruit in response to infection with three major postharvest fungi. Postharvest Biol. Technol. 2018, 146, 134–146. [Google Scholar] [CrossRef]
- Deising, H.B.; Gase, I.; Kubo, Y. The unpredictable risk imposed by microbial secondary metabolites: How safe is biological control of plant diseases? J. Plant Dis. Prot. 2017, 124, 413–419. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Early molecular events in PAMP-triggered immunity. Curr. Opin. Plant Biol. 2009, 12, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr. Opin. Plant Biol. 2018, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The Crosstalks Between Jasmonic Acid and Other Plant Hormone Signaling Highlight the Involvement of Jasmonic Acid as a Core Component in Plant Response to Biotic and Abiotic Stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, D.; Khurana, J.P. Role of pathogenesis-related (PR) proteins in plant defense mechanism. In Molecular Aspects of Plant-Pathogen Interaction; Springer: Berlin/Heidelberg, Germany, 2018; pp. 265–281. [Google Scholar]
- Wei, K.; Chen, J.; Wang, Y.; Chen, Y.; Chen, S.; Lin, Y.; Pan, S.; Zhong, X.; Xie, D. Genome-Wide Analysis of bZIP-Encoding Genes in Maize. DNA Res. 2012, 19, 463–476. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hao, J.; Chen, X.; Hao, Z.; Wang, X.; Lou, Y.; Peng, Y.; Guo, Z. Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Mol. Biol. 2007, 65, 799–815. [Google Scholar] [CrossRef]
- Jung, J.; Won, S.Y.; Suh, S.C.; Kim, H.; Wing, R.; Jeong, Y.; Hwang, I.; Kim, M. The barley ERF-type transcription factor HvRAF confers enhanced pathogen resistance and salt tolerance in Arabidopsis. Planta 2007, 225, 575–588. [Google Scholar] [CrossRef]
- Seo, P.J.; Kim, M.J.; Park, J.Y.; Kim, S.Y.; Jeon, J.; Lee, Y.H.; Kim, J.; Park, C.M. Cold activation of a plasma membrane-tethered NAC transcription factor induces a pathogen resistance response in Arabidopsis. Plant J. 2010, 61, 661–671. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Zhang, C.-L.; Wang, G.-L.; Wang, Y.-X.; Qi, C.-H.; Zhao, Q.; You, C.-X.; Li, Y.-Y.; Hao, Y.-J. The R2R3 MYB transcription factor MdMYB30 modulates plant resistance against pathogens by regulating cuticular wax biosynthesis. BMC Plant Biol. 2019, 19, 362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yao, X.; Ren, H.; Chang, J.; Wang, K. RNA-Seq reveals flavonoid biosynthesis-related genes in pecan (Carya illinoinensis) kernels. J. Agric. Food Chem. 2018, 67, 148–158. [Google Scholar] [CrossRef]
- Mattison, C.P.; Rai, R.; Settlage, R.E.; Hinchliffe, D.J.; Madison, C.; Bland, J.M.; Brashear, S.; Graham, C.J.; Tarver, M.R.; Florane, C. RNA-Seq analysis of developing pecan (Carya illinoinensis) embryos reveals parallel expression patterns among allergen and lipid metabolism genes. J. Agric. Food Chem. 2017, 65, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xi, D.; Chen, Y.; Zhu, C.; Zhao, Y.; Geng, G. Morphological characterization and transcriptome analysis of pistillate flowering in pecan (Carya illinoinensis). Sci. Hortic. 2019, 257, 108674. [Google Scholar] [CrossRef]
- Sa, R.; Liu, D.; Chen, L.; Liu, L.; Zhang, Y.; Zhang, X.; Xu, C. Transcriptome analysis of mechanisms and candidate genes associated with cucumber response to cucumber alternaria leaf spot infection. Physiol. Mol. Plant Pathol. 2020, 111, 101490. [Google Scholar] [CrossRef]
- Foster, A.J.; Pelletier, G.; Tanguay, P.; Seguin, A. Transcriptome analysis of poplar during leaf spot infection with Sphaerulina spp. PLoS ONE 2015, 10, e0138162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Shi, J.; Li, X.; Liu, J.; Geng, Q.; Shi, H.; Ke, Y.; Sun, Q. Transcriptome analysis reveals the molecular mechanisms of the defense response to gray leaf spot disease in maize. BMC Genom. 2018, 19, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, M.; Zhu, C.; Zhao, Y.; Wang, B.; Ma, X.; Geng, G. Field investigation of resistance against black spot of different pecan varieties in Jintan, Changzhou. J. Jiangsu For. Sci. Technol. 2018, 45, 26–29. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=JSLY201806007&DbName=CJFQ2018 (accessed on 17 December 2018).
- Walker, J.R.; Ferrar, P.H. Diphenol Oxidases, Enzyme-catalysed Browning and Plant Disease Resistance. Biotechnol. Genet. Eng. Rev. 1998, 15, 457–498. [Google Scholar] [CrossRef]
- Srivastava, S. Peroxidase and poly-phenol oxidase in Brassica juncea plants infected with Macrophomina phaseolina (Tassai) Goid. and their implication in disease resistance. J. Phytopathol. 1987, 120, 249–254. [Google Scholar] [CrossRef]
- Dowd, P.F.; Johnson, E.T. Association of a specific cationic peroxidase isozyme with maize stress and disease resistance responses, genetic identification, and identification of a cDNA coding for the isozyme. J. Agric. Food Chem. 2005, 53, 4464–4470. [Google Scholar] [CrossRef]
- Lurie, S.; Fallik, E.; Handros, A.; Shapira, R. The possible involvement of peroxidase in resistance to Botrytis cinereain heat treated tomato fruit. Physiol. Mol. Plant P 1997, 50, 141–149. [Google Scholar] [CrossRef]
- Seevers, P.; Daly, J.; Catedral, F. The role of peroxidase isozymes in resistance to wheat stem rust disease. Plant Physiol. 1971, 48, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.W.; Lee, B.G.; Kim, N.H.; Park, Y.; Lim, C.W.; Song, H.K.; Hwang, B.K. A role for a menthone reductase in resistance against microbial pathogens in plants. Plant Physiol. 2008, 148, 383–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herron, S.R.; Benen, J.A.E.; Scavetta, R.D.; Visser, J.; Jurnak, F. Structure and function of pectic enzymes: Virulence factors of plant pathogens. Proc. Natl. Acad. Sci. USA 2000, 97, 8762–8769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Zhang, S. MAPK Cascades in Plant Disease Resistance Signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Anderson, J.C.; Bartels, S.; Besteiro, M.A.G.; Shahollari, B.; Ulm, R.; Peck, S.C. Arabidopsis MAP Kinase Phosphatase 1 (AtMKP1) negatively regulates MPK6-mediated PAMP responses and resistance against bacteria. Plant J. 2011, 67, 258–268. [Google Scholar] [CrossRef]
- Ekengren, S.K.; Liu, Y.; Schiff, M.; Dinesh-Kumar, S.; Martin, G.B. Two MAPK cascades, NPR1, and TGA transcription factors play a role in Pto-mediated disease resistance in tomato. Plant J. 2003, 36, 905–917. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, L.; An, H.; Wu, C.; Guo, X. GhMPK16, a novel stress-responsive group D MAPK gene from cotton, is involved in disease resistance and drought sensitivity. BMC Mol. Biol. 2011, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Chen, J.; Song, F.; Zheng, Z. A novel rice MAPK gene, OsBIMK2, is involved in disease-resistance responses. Plant Biology 2006, 8, 587–596. [Google Scholar] [CrossRef]
- Patil, V.; Nandi, A.K. Role of MAPK Cascade in Local and Systemic Immunity of Plants. In Protein Kinases and Stress Signaling in Plants: Functional Genomic Perspective; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2020; pp. 422–444. [Google Scholar] [CrossRef]
- Schweighofer, A.; Meskiene, I. Regulation of stress hormones jasmonates and ethylene by MAPK pathways in plants. Mol. BioSyst. 2008, 4, 799–803. [Google Scholar] [CrossRef]
- Yu, F.-Q.; Han, C.-S.; Yang, W.; Jin, X.; Hu, Z.-Y.; Liu, Y.-X. Activation of the p38 MAPK pathway by follicle-stimulating hormone regulates steroidogenesis in granulosa cells differentially. J. Endocrinol. 2005, 186, 85–96. [Google Scholar] [CrossRef]
- Broekaert, W.F.; Delauré, S.L.; De Bolle, M.F.; Cammue, B.P. The Role of Ethylene in Host-Pathogen Interactions. Annu. Rev. Phytopathol. 2006, 44, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Browse, J. Jasmonate Passes Muster: A Receptor and Targets for the Defense Hormone. Annu. Rev. Plant Biol. 2009, 60, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Vlot, A.C.; Dempsey, D.M.A.; Klessig, D.F. Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.L.-C.; Li, H.; Ecker, J.R. Ethylene Biosynthesis and Signaling Networks. Plant Cell 2002, 14 (Suppl. 1), S131–S151. [Google Scholar] [CrossRef] [Green Version]
- Li, T.-T.; Zhang, J.-D.; Tang, J.-Q.; Liu, Z.-C.; Li, Y.-Q.; Chen, J.; Zou, L.-W. Combined use of Trichoderma atroviride CCTCCSBW0199 and brassinolide to control Botrytis cinerea infection in tomato. Plant Dis. 2020, 104, 1298–1304. [Google Scholar] [CrossRef]
- Kim, K.-C.; Lai, Z.; Fan, B.; Chen, Z. Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 2008, 20, 2357–2371. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Li, X.; Zheng, J. Transcriptome profiling using Illumina-and SMRT-based RNA-Seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene 2018, 666, 123–133. [Google Scholar] [CrossRef]
- Oh, S.-K.; Baek, K.-H.; Park, J.M.; Yi, S.Y.; Yu, S.H.; Kamoun, S.; Choi, D. Capsicum annuum WRKY protein CaWRKY1 is a negative regulator of pathogen defense. New Phytol. 2007, 177, 977–989. [Google Scholar] [CrossRef]
- Yang, B.; Jiang, Y.; Rahman, M.H.; Deyholos, M.K.; Kav, N.N. Identification and expression analysis of WRKY transcription factor genes in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biol. 2009, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Yu, X.; Chen, W.; Wang, S.; Cao, L.; Geng, X.; Sun, C.; Qu, S. Transcriptome sequencing, data-based screening, and functional investigation of MdWRKY75d and MdWRKY75e in disease-resistant apples. J. Plant Interact. 2021, 16, 462–473. [Google Scholar] [CrossRef]
- Zhou, R.; Dong, Y.; Liu, X.; Feng, S.; Wang, C.; Ma, X.; Liu, J.; Liang, Q.; Bao, Y.; Xu, S. JrWRKY21 interacts with JrPTI5L to activate expression of JrPR5L for resistance to Colletotrichum gloeosporioides in walnut. Plant J. 2022, 6, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Han, S.; He, D.; Cao, G.; Zhang, F.; Wan, X. Evaluating Walnut (Juglans spp.) for resistance to walnut blight and comparisons between artificial inoculation assays and field studies. Australas. Plant Pathol. 2019, 48, 221–231. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Wang, W.; Chen, Y.; Zhao, Y.; Zhang, S.; Shi, F.; Khalil-Ur-Rehman, M.; Nieuwenhuizen, N.J. Transcriptomics and Antioxidant Analysis of Two Chinese Chestnut (Castanea mollissima BL.) Varieties Provides New Insights Into the Mechanisms of Resistance to Gall Wasp Dryocosmus kuriphilus Infestation. Front. Plant Sci. 2022, 13, 874434. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Clean Reads (M) | Total Mapped Reads (%) | Unique Mapped Reads (%) | Clean Bases (Gb) | Clean Reads Q30 (%) |
|---|---|---|---|---|---|
| M_1 | 44.23 | 88.06 | 70.79 | 6.63 | 91.63 |
| M_2 | 43.53 | 86.66 | 68.62 | 6.53 | 91.01 |
| M_3 | 40.76 | 86.57 | 68.57 | 6.11 | 91.12 |
| W_1 | 45.46 | 84.11 | 65.08 | 6.82 | 91.03 |
| W_2 | 44.14 | 84.33 | 65.45 | 6.62 | 91.16 |
| W_3 | 42.22 | 86.17 | 66.87 | 6.33 | 90.98 |
| X4_1 | 44.18 | 85.25 | 66.21 | 6.63 | 90.81 |
| X4_2 | 40.6 | 87.72 | 69.71 | 6.09 | 91.94 |
| X4_3 | 45.35 | 86.76 | 68.64 | 6.8 | 91.95 |
| Pathway ID | Pathway | DEGs with Pathway Annotation (log2 (Fold-Change) > 1) | p-Value (<0.05) | ||
|---|---|---|---|---|---|
| Total | Upregulated | Downregulated | |||
| M vs. X4 | |||||
| ko04626 | Plant–pathogen interaction | 249 | 116 | 133 | 5.66 × 10−16 |
| ko00195 | Photosynthesis | 29 | 28 | 1 | 1.61 × 10−9 |
| ko04016 | MAPK signaling pathway, plant | 155 | 76 | 79 | 9.28 × 10−8 |
| ko00500 | Starch and sucrose metabolism | 150 | 79 | 71 | 2.64 × 10−7 |
| ko00051 | Fructose and mannose metabolism | 43 | 25 | 18 | 3.74 × 10−7 |
| ko00196 | Photosynthesis, antenna proteins | 11 | 11 | 0 | 2.16 × 10−5 |
| ko00965 | Betalain biosynthesis | 12 | 12 | 0 | 2.80 × 10−5 |
| ko00901 | Indole alkaloid biosynthesis | 13 | 13 | 0 | 3.02 × 10−5 |
| ko04075 | Plant hormone signal transduction | 139 | 75 | 64 | 0.000291 |
| ko00030 | Pentose phosphate pathway | 41 | 23 | 18 | 0.000543 |
| ko00592 | Alpha-linolenic acid metabolism | 27 | 11 | 16 | 0.006199 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 30 | 17 | 13 | 0.01525 |
| ko01040 | Biosynthesis of unsaturated fatty acids | 15 | 5 | 10 | 0.039254 |
| W vs. M | |||||
| ko04016 | MAPK signaling pathway, plant | 236 | 60 | 176 | 1.32 × 10−8 |
| ko04075 | Plant hormone signal transduction | 223 | 106 | 117 | 1.43 × 10−5 |
| ko01040 | Biosynthesis of unsaturated fatty acids | 24 | 10 | 14 | 0.011575 |
| ko04626 | Plant–pathogen interaction | 290 | 84 | 206 | 0.011575 |
| W vs. X4 | |||||
| ko04016 | MAPK signaling pathway, plant | 489 | 162 | 327 | 4.37 × 10−24 |
| ko04626 | Plant–pathogen interaction | 651 | 238 | 413 | 2.04 × 10−16 |
| ko00196 | Photosynthesis, antenna proteins | 23 | 23 | 0 | 6.00 × 10−10 |
| ko04075 | Plant hormone signal transduction | 421 | 230 | 191 | 7.61 × 10−9 |
| ko00195 | Photosynthesis | 48 | 46 | 2 | 1.55 × 10−6 |
| ko00592 | Alpha-linolenic acid metabolism | 78 | 25 | 53 | 3.50 × 10−6 |
| ko01040 | Biosynthesis of unsaturated fatty acids | 44 | 15 | 29 | 8.08 × 10−5 |
| ko00564 | Glycerophospholipid metabolism | 107 | 60 | 47 | 0.001253 |
| ko00051 | Fructose and mannose metabolism | 77 | 52 | 25 | 0.024548 |
| ko00965 | Betalain biosynthesis | 16 | 16 | 0 | 0.024742 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhang, S.; Zhao, Y.; Mo, Z.; Wang, W.; Zhu, C. Transcriptomic Analysis to Unravel Potential Pathways and Genes Involved in Pecan (Carya illinoinensis) Resistance to Pestalotiopsis microspora. Int. J. Mol. Sci. 2022, 23, 11621. https://doi.org/10.3390/ijms231911621
Chen Y, Zhang S, Zhao Y, Mo Z, Wang W, Zhu C. Transcriptomic Analysis to Unravel Potential Pathways and Genes Involved in Pecan (Carya illinoinensis) Resistance to Pestalotiopsis microspora. International Journal of Molecular Sciences. 2022; 23(19):11621. https://doi.org/10.3390/ijms231911621
Chicago/Turabian StyleChen, Yu, Shijie Zhang, Yuqiang Zhao, Zhenghai Mo, Wu Wang, and Cancan Zhu. 2022. "Transcriptomic Analysis to Unravel Potential Pathways and Genes Involved in Pecan (Carya illinoinensis) Resistance to Pestalotiopsis microspora" International Journal of Molecular Sciences 23, no. 19: 11621. https://doi.org/10.3390/ijms231911621