
TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β

 ,
,  , ,
, ,  , , ,
, , , 
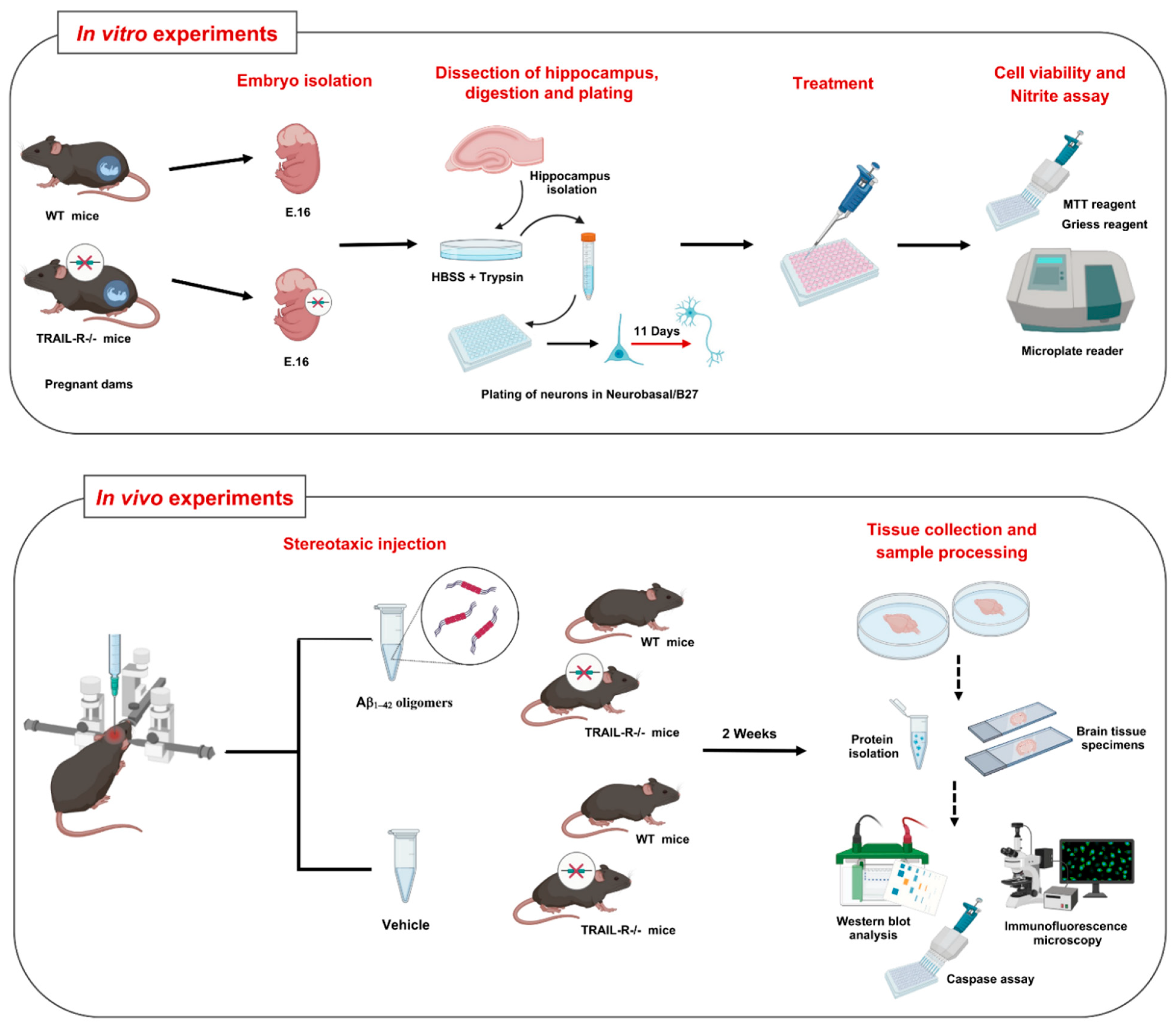{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Amyloid Beta Neurotoxicity Is Significantly Attenuated in TRAIL-R−/− Mouse Primary Neuronal Cells
2.2. TRAIL-R2 Is Required for p53 to Mediate Aβ-Related Neurotoxicity
2.3. TRAIL-R−/− Mice Show Reduced Caspase Activity after Challenge with Aβ1-42
2.4. JNK and AKT Kinases Are Inversely Modulated in TRAIL-R−/− Mice That Have Undergone Oligomeric Aβ1-42 Treatment
2.5. Aβ1-42 Dependent GSK3β Activation and Tau Phosphorylation Are Attenuated in TRAIL-R−/− Mice
2.6. Glial Response Is Blunted in TRAIL-R−/− Mice Treated with Oligomeric Aβ1-42
2.7. Inflammatory Molecules Expression Is Reduced in TRAIL-R−/− Mice Receiving Oligomeric Aβ1-42
2.8. Nitrite Levels Are Significantly Attenuated in the Media from Embryonic Hippocampal Cell Cultures from TRAIL-R−/− Mice Treated with Oligomeric Aβ1-42
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Preparation of Aβ1-42 Oligomers
4.3. Western Blot Analysis of Aβ1–42 Oligomers
4.4. Experimental Groups and Drug Administration
4.5. Primary Cultures of Mouse Hippocampal Neurons
4.6. Cell Viability Assay
4.7. Free-Floating Fluorescence Immunohistochemistry
4.8. Protein Extraction
4.9. Western Blot Analysis
4.10. Caspase Colorimetric Assay
4.11. Nitrite Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coupé, P.; Manjón, J.V.; Lanuza, E.; Catheline, G. Lifespan Changes of the Human Brain In Alzheimer’s Disease. Sci. Rep. 2019, 9, 3998. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s Disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic Synaptic Plasticity and Dysfunction in Alzheimer Disease: Emerging Mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate Immunity in Alzheimer’s Disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- Cao, W.; Zheng, H. Peripheral Immune System in Aging and Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- McAlpine, F.E.; Tansey, M.G. Neuroinflammation and Tumor Necrosis Factor Signaling in the Pathophysiology of Alzheimer’s Disease. J. Inflamm. Res. 2008, 1, 29–39. [Google Scholar] [CrossRef][Green Version]
- Cantarella, G.; Di Benedetto, G.; Puzzo, D.; Privitera, L.; Loreto, C.; Saccone, S.; Giunta, S.; Palmeri, A.; Bernardini, R. Neutralization of TNFSF10 Ameliorates Functional Outcome in a Murine Model of Alzheimer’s Disease. Brain 2015, 138, 203–216. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Saccone, S.; Lempereur, L.; Ronsisvalle, N.; Nocentini, G.; Bianchini, R.; Riccardi, C.; Bernardini, R.; Cantarella, G. The Proinflammatory Cytokine GITRL Contributes to TRAIL-Mediated Neurotoxicity in the HCN-2 Human Neuronal Cell Line. Curr. Alzheimer Res. 2017, 14, 1090–1101. [Google Scholar] [CrossRef]
- Cantarella, G.; Lempereur, L.; D’Alcamo, M.A.; Risuglia, N.; Cardile, V.; Pennisi, G.; Scoto, G.M.; Bernardini, R. Trail Interacts Redundantly with Nitric Oxide in Rat Astrocytes: Potential Contribution to Neurodegenerative Processes. J. Neuroimmunol. 2007, 182, 41–47. [Google Scholar] [CrossRef]
- Ryan, L.A.; Peng, H.; Erichsen, D.A.; Huang, Y.; Persidsky, Y.; Zhou, Y.; Gendelman, H.E.; Zheng, J. TNF-Related Apoptosis-Inducing Ligand Mediates Human Neuronal Apoptosis: Links to HIV-1-Associated Dementia. J. Neuroimmunol. 2004, 148, 127–139. [Google Scholar] [CrossRef]
- Huang, Y.; Erdmann, N.; Peng, H.; Zhao, Y.; Zheng, J. The Role of TNF Related Apoptosis-Inducing Ligand in Neurodegenerative Diseases. Cell. Mol. Immunol. 2005, 2, 113–122. [Google Scholar]
- Martin-Villalba, A.; Herr, I.; Jeremias, I.; Hahne, M.; Brandt, R.; Vogel, J.; Schenkel, J.; Herdegen, T.; Debatin, K.M. CD95 Ligand (Fas-L/APO-1L) and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Mediate Ischemia-Induced Apoptosis in Neurons. J. Neurosci. 1999, 19, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Pignataro, G.; Di Benedetto, G.; Anzilotti, S.; Vinciguerra, A.; Cuomo, O.; Di Renzo, G.F.; Parenti, C.; Annunziato, L.; Bernardini, R. Ischemic Tolerance Modulates TRAIL Expression and Its Receptors and Generates a Neuroprotected Phenotype. Cell Death Dis. 2014, 5, e1331. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Di Benedetto, G.; Scollo, M.; Paterniti, I.; Cuzzocrea, S.; Bosco, P.; Nocentini, G.; Riccardi, C.; Bernardini, R. Neutralization of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Reduces Spinal Cord Injury Damage in Mice. Neuropsychopharmacology 2010, 35, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Uberti, D.; Carsana, T.; Lombardo, G.; Bernardini, R.; Memo, M. Neutralization of TRAIL Death Pathway Protects Human Neuronal Cell Line from β-Amyloid Toxicity. Cell Death Differ. 2003, 10, 134–141. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and Its Death and Decoy Receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A Novel Apoptosis-Mediating Receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular Cloning and Functional Analysis of the Mouse Homologue of the KILLER/DR5 Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Death Receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar] [PubMed]
- Dörr, J.; Bechmann, I.; Waiczies, S.; Aktas, O.; Walczak, H.; Krammer, P.H.; Nitsch, R.; Zipp, F. Lack of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand but Presence of Its Receptors in the Human Brain. J. Neurosci. 2002, 22, RC209. [Google Scholar] [CrossRef] [PubMed]
- Uberti, D.; Cantarella, G.; Facchetti, F.; Cafici, A.; Grasso, G.; Bernardini, R.; Memo, M. TRAIL Is Expressed in the Brain Cells of Alzheimer’s Disease Patients. Neuroreport 2004, 15, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Uberti, D.; Ferrari-Toninelli, G.; Bonini, S.A.; Sarnico, I.; Benarese, M.; Pizzi, M.; Benussi, L.; Ghidoni, R.; Binetti, G.; Spano, P.; et al. Blockade of the Tumor Necrosis Factor-Related Apoptosis Inducing Ligand Death Receptor DR5 Prevents β-Amyloid Neurotoxicity. Neuropsychopharmacology 2007, 32, 872–880. [Google Scholar] [CrossRef]
- Finnberg, N.; Klein-Szanto, A.J.P.; El-Deiry, W.S. TRAIL-R Deficiency in Mice Promotes Susceptibility to Chronic Inflammation and Tumorigenesis. J. Clin. Investig. 2008, 118, 111–123. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Khuri, F.R.; Sun, S.-Y. Decoy Receptor 2 (DcR2) Is a P53 Target Gene and Regulates Chemosensitivity. Cancer Res. 2005, 65, 9169–9175. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Fornace, A.J. Death and Decoy Receptors and P53-Mediated Apoptosis. Leukemia 2000, 14, 1509–1513. [Google Scholar] [CrossRef]
- Ruiz de Almodóvar, C.; Ruiz-Ruiz, C.; Rodríguez, A.; Ortiz-Ferrón, G.; Redondo, J.M.; López-Rivas, A. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Decoy Receptor TRAIL-R3 Is up-Regulated by P53 in Breast Tumor Cells through a Mechanism Involving an Intronic P53-Binding Site. J. Biol. Chem. 2004, 279, 4093–4101. [Google Scholar] [CrossRef]
- Toscano, F.; Fajoui, Z.E.; Gay, F.; Lalaoui, N.; Parmentier, B.; Chayvialle, J.-A.; Scoazec, J.-Y.; Micheau, O.; Abello, J.; Saurin, J.-C. P53-Mediated Upregulation of DcR1 Impairs Oxaliplatin/TRAIL-Induced Synergistic Anti-Tumour Potential in Colon Cancer Cells. Oncogene 2008, 27, 4161–4171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailey, S.L.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schütz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R Deficiency in Mice Enhances Lymph Node Metastasis without Affecting Primary Tumor Development. J. Clin. Investig. 2008, 118, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Diehl, G.E.; Yue, H.H.; Hsieh, K.; Kuang, A.A.; Ho, M.; Morici, L.A.; Lenz, L.L.; Cado, D.; Riley, L.W.; Winoto, A. TRAIL-R as a Negative Regulator of Innate Immune Cell Responses. Immunity 2004, 21, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.S.; Kinoshita, Y. The Role of P53 in Neuronal Cell Death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Hall, C.K.; Ngo, L.; Jay, G. Extracellular Deposition of Beta-Amyloid upon P53-Dependent Neuronal Cell Death in Transgenic Mice. J. Clin. Investig. 1996, 98, 1626–1632. [Google Scholar] [CrossRef]
- Szybińska, A.; Leśniak, W. P53 Dysfunction in Neurodegenerative Diseases—The Cause or Effect of Pathological Changes? Aging Dis. 2017, 8, 506–518. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Ashkenazi, A. New Insights into Apoptosis Signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef]
- Cantarella, G.; Di Benedetto, G.; Pezzino, S.; Risuglia, N.; Bernardini, R. TRAIL-Related Neurotoxicity Implies Interaction with the Wnt Pathway in Human Neuronal Cells in Vitro. J. Neurochem. 2008, 105, 1915–1923. [Google Scholar] [CrossRef]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.a.E. Non-Canonical Kinase Signaling by the Death Ligand TRAIL in Cancer Cells: Discord in the Death Receptor Family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-Signaling: A Multiplexing Hub in Programmed Cell Death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen Synthase Kinase-3 Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Boje, K.M.K. Nitric Oxide Neurotoxicity in Neurodegenerative Diseases. Front. Biosci. 2004, 9, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Asiimwe, N.; Yeo, S.G.; Kim, M.-S.; Jung, J.; Jeong, N.Y. Nitric Oxide: Exploring the Contextual Link with Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2016, 2016, 7205747. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Wu, G.S.; Burns, T.F.; McDonald, E.R.; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 Is a DNA Damage-Inducible P53-Regulated Death Receptor Gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Willms, A.; Schupp, H.; Poelker, M.; Adawy, A.; Debus, J.F.; Hartwig, T.; Krichel, T.; Fritsch, J.; Singh, S.; Walczak, H.; et al. TRAIL-Receptor 2-a Novel Negative Regulator of P53. Cell Death Dis. 2021, 12, 757. [Google Scholar] [CrossRef]
- Finnberg, N.; Gruber, J.J.; Fei, P.; Rudolph, D.; Bric, A.; Kim, S.-H.; Burns, T.F.; Ajuha, H.; Page, R.; Wu, G.S.; et al. DR5 Knockout Mice Are Compromised in Radiation-Induced Apoptosis. Mol. Cell. Biol. 2005, 25, 2000–2013. [Google Scholar] [CrossRef]
- Ronsisvalle, N.; Di Benedetto, G.; Parenti, C.; Amoroso, S.; Bernardini, R.; Cantarella, G. CHF5074 Protects SH-SY5Y Human Neuronal-like Cells from Amyloidbeta 25–35 and Tumor Necrosis Factor Related Apoptosis Inducing Ligand Toxicity in Vitro. Curr. Alzheimer Res. 2014, 11, 714–724. [Google Scholar] [CrossRef]
- Magrané, J.; Rosen, K.M.; Smith, R.C.; Walsh, K.; Gouras, G.K.; Querfurth, H.W. Intraneuronal Beta-Amyloid Expression Downregulates the Akt Survival Pathway and Blunts the Stress Response. J. Neurosci. 2005, 25, 10960–10969. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting Neuroinflammation in Alzheimer’s Disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef]
- Wang, S.; Yang, H.; Yu, L.; Jin, J.; Qian, L.; Zhao, H.; Xu, Y.; Zhu, X. Oridonin Attenuates Aβ1–42-Induced Neuroinflammation and Inhibits NF-ΚB Pathway. PLoS ONE 2014, 9, e104745. [Google Scholar] [CrossRef]
- Wu, J.; Wang, A.; Min, Z.; Xiong, Y.; Yan, Q.; Zhang, J.; Xu, J.; Zhang, S. Lipoxin A4 Inhibits the Production of Proinflammatory Cytokines Induced by β-Amyloid in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2011, 408, 382–387. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Carta, A.R.; Saccone, S.; Lempereur, L.; Mulas, G.; Loreto, C.; Bernardini, R.; Cantarella, G. Beneficial Effects of Curtailing Immune Susceptibility in an Alzheimer’s Disease Model. J. Neuroinflammation 2019, 16, 166. [Google Scholar] [CrossRef]
- Fa, M.; Orozco, I.J.; Francis, Y.I.; Saeed, F.; Gong, Y.; Arancio, O. Preparation of Oligomeric β-Amyloid1-42 and Induction of Synaptic Plasticity Impairment on Hippocampal Slices. J. Vis. Exp. 2010, 41, e1884. [Google Scholar] [CrossRef]
- Jean, Y.Y.; Baleriola, J.; Fà, M.; Hengst, U.; Troy, C.M. Stereotaxic Infusion of Oligomeric Amyloid-beta into the Mouse Hippocampus. J. Vis. Exp. 2015, 100, e52805. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Benedetto, G.; Burgaletto, C.; Serapide, M.F.; Caltabiano, R.; Munafò, A.; Bellanca, C.M.; Di Mauro, R.; Bernardini, R.; Cantarella, G. TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β. Int. J. Mol. Sci. 2022, 23, 11625. https://doi.org/10.3390/ijms231911625
Di Benedetto G, Burgaletto C, Serapide MF, Caltabiano R, Munafò A, Bellanca CM, Di Mauro R, Bernardini R, Cantarella G. TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β. International Journal of Molecular Sciences. 2022; 23(19):11625. https://doi.org/10.3390/ijms231911625
Chicago/Turabian StyleDi Benedetto, Giulia, Chiara Burgaletto, Maria Francesca Serapide, Rosario Caltabiano, Antonio Munafò, Carlo Maria Bellanca, Rosaria Di Mauro, Renato Bernardini, and Giuseppina Cantarella. 2022. "TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β" International Journal of Molecular Sciences 23, no. 19: 11625. https://doi.org/10.3390/ijms231911625
APA StyleDi Benedetto, G., Burgaletto, C., Serapide, M. F., Caltabiano, R., Munafò, A., Bellanca, C. M., Di Mauro, R., Bernardini, R., & Cantarella, G. (2022). TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β. International Journal of Molecular Sciences, 23(19), 11625. https://doi.org/10.3390/ijms231911625