Abstract
A number of bacteria that colonize the human body produce toxins and effectors that cause changes in the eukaryotic cell cycle—cyclomodulins and low-molecular-weight compounds such as butyrate, lactic acid, and secondary bile acids. Cyclomodulins and metabolites are necessary for bacteria as adaptation factors—which are influenced by direct selection—to the ecological niches of the host. In the process of establishing two-way communication with the macroorganism, these compounds cause limited damage to the host, despite their ability to disrupt key processes in eukaryotic cells, which can lead to pathological changes. Possible negative consequences of cyclomodulin and metabolite actions include their potential role in carcinogenesis, in particular, with the ability to cause DNA damage, increase genome instability, and interfere with cancer-associated regulatory pathways. In this review, we aim to examine cyclomodulins and bacterial metabolites as important factors in bacterial survival and interaction with the host organism to show their heterogeneous effect on oncogenesis depending on the surrounding microenvironment, pathological conditions, and host genetic background.
1. Introduction
Epidemiological studies often reveal strong links between bacterial pathogens and cancer incidence: between Helicobacter pylori and gastric cancer; between Salmonella typhi and gallbladder carcinoma; between Salmonella enteritidis and colon carcinoma. Therein, many bacterial factors have been identified that mediate the malignant transformation of host cells: toxins, cell surface components, and effector proteins. Frequently, these factors target eukaryotic cell signaling pathways; interference with these pathways leads to cancer development [1]. Thus, many bacterial pathogens affect the Wnt/β-catenin signaling pathway, resulting in the induction of the β-catenin release. β-catenin penetrates the nucleus and activates the Wnt pathway genes to control the transcription of genes involved in apoptosis, cell proliferation, and malignant transformation [2,3]. Bacterial surface molecules can perform as activators, for example, Fusobacterium nucleatum adhesin FadA, which can bind to E-cadherin, inducing the release of β-catenin [4]. The S. enteritidis AvrA effector protein stabilizes β-catenin, which leads to Wnt signaling, and inhibits JNK and NF-κβ signaling pathways involved in inflammation and apoptosis [1]. The CagA toxin of H. pylori forms a functional CagA-c-Met-CD44 complex by activating Wnt/β-catenin, which induces the accumulation of nuclear β-catenin by activating PI3K/Akt signaling, resulting in the inhibition of apoptosis. Bacteroides fragilis toxin (BFT) causes dissociation of β-catenin from E-cadherin; the complex of β-catenin with TCF4 leads to c-Myc expression and cell proliferation [3]. No less significant bacterial factors associated with the malignant transformation of eukaryotic cells are toxins and effectors that cause changes in the eukaryotic cell cycle, for which they received their name—cyclomodulins. The cell cycle is one of the most favorable targets for toxins and effectors because basic cell functions are disrupted, which creates favorable conditions for bacterial invasion and colonization of the host. Cell cycle arrest at the G2/M phase by some cyclomodulins leads to disruption of cell renewal, induces delayed apoptosis of host cells, or blocks it. This increases the bacterial replication time and promotes colonization of the host, but at the same time can lead the cells to malignant transformation. Cyclomodulins of many commensal bacteria induce cell cycle arrest at the G2/M phase, creating a favorable environment for themselves without causing significant harm to the host [5].
The ecology of bacteria occupying the niches of mucous membranes and skin is associated with the colonization of these surfaces by commensals and pathogens, which, in the course of a long evolution, have developed various survival strategies both in the microbial environment and in the macro-organism [6]. Cyclomodulins are directly related to one of the survival strategies of bacteria. This strategy is based on causing limited damage to host cells, which disrupts their normal functions and defense against the pathogenic potential of the bacteria themselves. [7,8]. These cyclomodulins include cytotoxic necrotizing factors (CNFs), which prevent cytokinesis and simultaneously stimulate the cell cycle and DNA replication; cycle inhibitory factors (CIFs) that block the cell cycle and slow down apoptosis by inhibiting the proteasomal degradation pathway; DNA-damaging genotoxins, including a low-molecular-weight substance colibactin and protein cytolethal distending toxin (CDT) [9,10,11,12]. By modulating cell differentiation, apoptosis, and proliferation, cyclomodulins slow down the renewal of the epithelium, contributing to the prolonged colonization of macroorganism biotopes by the bacteria producing them. At the same time, eukaryotic cells preserved after exposure to toxins acquire genomic instability, which can play a certain role in their malignant transformation [13,14].
Unlike bacteria that adapt to the host through the production of toxins affecting its cells, there is a different microbial survival strategy—inhibition of competing microorganisms with the help of microbial metabolites. Bacteria producers of a number of known compounds, such as butyrate, lactic acid and secondary bile acids, colonize various biotopes of the human body supporting different physiological processes. However, under conditions of disruption of the homeostasis of the macroorganism by external and internal factors, microbial metabolism can affect the occurrence and/or development of pathological conditions, including neoplasms [15]. Among the carcinogenic metabolites of bacteria, the most studied are hydrogen sulfide and secondary bile acids, as well as reactive oxygen intermediates capable of causing oxidative damage to DNA. While the role of secondary bile acids in carcinogenesis is known, the role of butyrate, lactate, and other derivatives of short-chain fatty acids is described mainly as protective against cancer development [3]. At the same time, studies emerge that prove their contribution to carcinogenesis.
Strategies have evolved as microorganisms have adapted to specific host niches, leading to selective advantages and efficient survival of the microbes that acquire them. Despite the fact that the implementation of these strategies causes minor damage to the host and is associated only with the possibility of the spread of microorganisms, the human microbiota is increasingly considered an inducer or modulator of pathological processes, including many forms of cancer [16]. Most of the human population is colonized by tumor-associated microorganisms, but their presence does not mean that disease will develop in the future. Large-scale epidemiological studies are required to prove the carcinogenic effect of a large number of microorganisms with oncogenic potential. In most studies, the role of microorganisms in carcinogenesis is assessed experimentally in vitro and in vivo and based on the analysis of the microbiome of patients with cancer compared with healthy people. The most studied in the context of association with oncological diseases is the intestinal microbiota. [17,18,19].
Given the accumulated data on potential carcinogenic bacteria and their metabolic products, it is assumed that the manifestation of the carcinogenic properties of microbial toxins and metabolites depends on a combination of factors, such as the host’s genetic predisposition to cancer, chronic inflammatory processes, and the influence of the bio-environment [20].
In this review, we sought to understand the role of cyclomodulins and bacterial metabolites as factors in the survival and adaptation of bacteria to the host organism. Having assessed the positive role of these factors for the producing bacteria themselves, we have investigated possible changes in the host organism, in which the impact of cyclomodulins and microbial metabolites leads to the development of cancer. Finally, we have outlined the mechanisms and causes of tumor development and/or progression when eukaryotic cells are exposed to these compounds in vitro and in vivo.
2. Cyclomodulins—CNFs
CNFs are bacterial exotoxins of a protein nature that affect signal transduction in a eukaryotic cell, modulating cytokinetic processes. CNFs activate GTP-binding proteins of the Rho family (Rho GTPases): Rho (A, B, C) Rac, Cdc42 in cells via deamidation of glutamine (Q61 or Q63) at the Rho active site; as a result, Rho proteins lose the ability to hydrolyze the Rho-bound GTP [21,22]. Activation of Rho GTPase proteins blocks key regulators of cellular processes in their active state, causing changes in the actin cytoskeleton, cell proliferation, production of reactive oxygen species (ROS), and release of anti-apoptotic and pro-inflammatory factors [23,24].
CNF toxins are secreted and bind to eukaryotic cell surface receptors, causing endocytosis and subsequent translocation of the catalytic subunit into the cytosol. CNFs were found in Escherichia coli, Yersinia pseudotuberculosis, Shigella species, and Salmonella enterica. CNF1 is the most studied toxin produced by some strains of E. coli and is more common among uropathogenic E. coli living in the intestine and penetrating into the urinary tract through the urethra [9,22,25].
CNF toxins activate Rho GTPases, which, through the Akt/IkB kinase pathway, positively regulate the activity of NFkB, which in turn translocates from the cytoplasm to the nucleus and promotes transcription of the anti-apoptotic factors Bcl-2 and Bcl-XL and cell survival [7,26]. Protecting epithelial cells from apoptosis, CNF1 slows down their renewal, which leads to the colonization of the epithelium by toxigenic bacteria. Other important events that occur under the influence of toxin-activated Rho GTPases are changes in the actin cytoskeleton through actin polymerization and blocking of cytokinesis with the formation of multinucleated cells as a result of ongoing DNA replication. Infected cells develop lamellipodia and filopodia, endowing them with phagocytic behavior and the ability to macropinocytosis, facilitating the internalization of bacteria into host cells [23,27].
CNF1′s potential role in carcinogenesis is also determined by its effect on eukaryotic cells (Figure 1). The absence of cytokinesis results in aneuploidy, which has been demonstrated by the treatment of the HCT 116 cell line with CNF1 toxin, the cell cycle reversibly arrested, and cell depolyploidization occurred. Their further entry into the cell cycle generated a large number of aneuploid progeny, which became more resistant to CNF1 [11]. Intercellular junctions disrupted under the influence of CNF1 improved the motility and migration of tumor cells, which can enhance their invasion and metastasis [23].
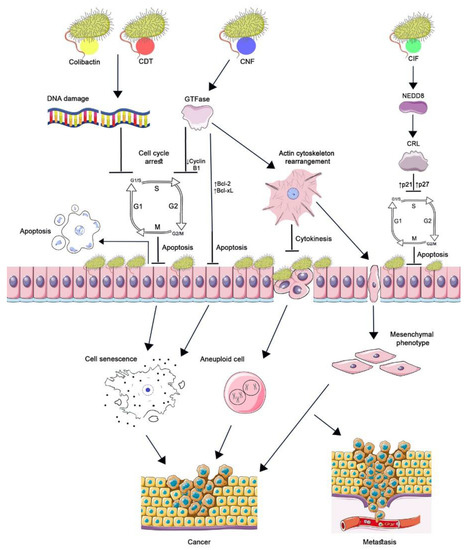
Figure 1.
The role of cyclomodulins in carcinogenesis. CNF1, activating Rho GTPases, causes changes in the actin cytoskeleton, which leads to blocking of cytokinesis and the occurrence of aneuploidy and the epithelial-mesenchymal transition of cells. GTPases suppress the expression of cyclin B1 by arresting the cell cycle in the G2/M phase and increasing the transcription of Bcl-2 and Bcl-XL factors without triggering apoptosis. Aneuploidy, senescence, and the acquisition of a mesenchymal phenotype by cells lead to the development and progression of cancer. CIF, by binding to the ubiquitin-like protein NEDD8, inhibits the activity of CRL ubiquitin ligase. As a result, p21 and p27 accumulate in the cell, which leads to cell cycle arrest, prevention of eukaryotic cell proliferation, and delayed apoptosis. The genotoxins colibactin and CDT cause DNA damage resulting in cell cycle arrest at the G1/S and G2/M checkpoints. Cell growth and cell renewal arrest facilitate bacterial colonization. The cellular response to significant DNA damage consists either in the development of apoptosis or in the formation of a cellular senescence phenotype. Incomplete DNA repair of surviving cells leads to genomic instability, initiation of tumor development, and/or increased tumor growth. (Figure 1 was created using the images from Servier Medical Art https://smart.servier.com/ 26 June 2022).
An experimentally in vitro epithelial-mesenchymal transition of intestinal epithelial cells induced by CNF1 was established. The acquisition of a mesenchymal phenotype by cells is currently recognized as one of the decisive factors in cancer progression [28]. It was also evident that GTPases activated by the Rho toxin can suppress the expression of cyclin B1; as a result, tumor cells (HeLa) are blocked in the G2/M phase without triggering an apoptotic response [29].
Despite the supposed role of the toxin in the development and/or progression of tumors, there are also records of the antitumor effect of CNF1 on mouse and human glioma cells, which leads to the blocking of cytokinesis of proliferating tumor cells, their aging, and death [30]. A recombinant CTX-CNF1 toxin that can penetrate the blood–brain barrier, selectively recognizes and affects glioma cells, and was created based on CNF1 and chlorotoxin (CTX) obtained from the venom of the scorpion Leiurus quinquestriatus [31].
Despite the specific activity of CNF1 against Rho GTPases and their permanent activation as a result of the constitutive association of Rho GTPase with GTP, some of the Rho pathways are activated temporarily. In this case, Rho proteins are depleted as a result of their ubiquitin-mediated proteasome degradation, which depends on the cell type. Thus, in HUVEC endothelial cells, macrophages, keratinocytes, fibroblasts, and 804G epithelial cells, the level of Rho ubiquitination is high, while in HEp-2, Vero, and HEK293 cells, it is low [26].
3. Cyclomodulins—CIFs
CIFs belong to bacterial cyclomodulins with enzymatic activities. The injection of CIF into the eukaryotic cell is carried out by the type III secretion system (T3SS), which transports various effector proteins into the host cell. CIF contributes to changes in eukaryotic cellular pathways in favor of the pathogen by arresting the cell cycle in the G1/S and G2/M phases. Irreversible cytopathic effects of CIF were first described in enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC) in an infection model on cultured HeLa cells [32].
CIF homologs were found in other bacteria of vertebrates (e.g., Burkholderia pseudomallei and Y. pseudotuberculosis) and invertebrates (e.g., Photorhabdus luminescens and Photorhabdus asymbiotica), and it has been shown that CIFs are conserved cyclomodulin proteins due to the presence of a conserved catalytic triad (Cys109-His165-Gln185). When the residues of this triad mutate, the cytopathic effect of CIF is not manifested [32,33].
CIF is encoded by a lambda-like prophage, and the cif gene may have spread widely through horizontal gene transfer. It is assumed that prophages encoding CIF are positively selected in bacteria to give them certain advantages in realizing their pathogenic potential. Thus, CIF in EPEC and EHEC is closely related to the LEE (locus of enterocyte effacement) pathogenicity island. CIF uses the existing T3SS, encoded by the LEE pathogenicity island, to be injected into target cells, and the resulting cytopathic effects slow down the turnover of affected cells. This gives selective advantages to LEE-positive E. coli over competing bacteria, especially in short-term adaptation processes [34].
Cell cycle arrest in the G2/M phase is associated with sustained CDK1 phosphorylation induced by CIF. At the same time, CIF is not a genotoxin and does not damage DNA like other cyclomodulins such as CDT or colibactin; therefore, it induces G2/M transition arrest regardless of activation of the DNA damage checkpoint pathway. CIF also blocks the entry of eukaryotic cells into the S phase, stopping the cell cycle in the G1/S phase. As a result of CIF action, CDK inhibitor proteins accumulate in cells: p21 inhibits the CDK1/CyclinB complex and the G2/M transition; p27, which, along with p21, inhibits the CDK2/CyclinE and A complexes and the G1/S transition [12,35].
Further studies have shown that the accumulation of p21 and p27 in infected cells is associated with the inactivation of CRL (Cullin-Ring ubiquitin Ligase), in which p21 and p27 are substrates of, and are degraded by, the ubiquitin-proteasome system. CIF, having deamidase enzymatic activity against NEDD8, a ubiquitin-like protein, impairs the conjugation of NEDD8 to Cullin, which leads to CRL inactivation. Thus, CIF inhibits ubiquitin-dependent degradation pathways of p21 and p27 proteins that regulate the cell cycle (Figure 1) [35,36].
The end result of inhibition of the proteasomal degradation pathway is cell cycle arrest prior to host cell destruction and apoptosis. By preventing proliferation, CIF can inhibit intestinal epithelial cell turnover, enhancing bacterial colonization. Delayed apoptosis or inhibition of apoptosis, associated, for example, with the accumulation of p21 in cells, also contributes to the local persistence of microorganisms. In addition, in an in vitro experiment, the time of death of cells exposed to CIF differed in various cell lines, which depended on the genetic background of the cells: HeLa cells died 72 h after infection; intestinal epithelial cells (IEC-6) with positive p53 died 48 h after infection [12,33].
Although, in in vitro experiments, CIF inhibited the proliferation of cancer cells and eventually led to apoptosis, where new epidemiological data confirm the role of CIF in carcinogenesis. In a recent multicenter case-control study, an association of the cif gene with precancerous lesions—intestinal polyps or adenomas—was found that can occur in the early stages of carcinogenesis, and this association has been assessed as a statistically significant risk factor [37].
4. Cyclomodulins—Genotoxins
Among the bacterial toxins, there is a unique group of genotoxins whose molecular target is DNA. The action of these toxins implements one of the strategies for the survival and adaptation of microorganisms to the ecological niche, aimed at causing damage to the host cells. Currently, three bacterial toxins are classified as genotoxins: polyketide toxin (colibactin), cytolethal distending toxin, and a toxin produced by S. typhi [38,39].
All eukaryotic cells respond to DNA damage and repair it, preserving the integrity of the genome. DNA breaks not eliminated during repair lead to cytogenetic disorders, oncotransformation, or cell death. Genotoxins cause single-strand or double-strand DNA breaks in target cells and trigger a signaling pathway that prevents the transition between phases of the cell cycle, resulting in its arrest, cellular senescence, or apoptosis [20,38].
Inhibition of cell proliferation and renewal by genotoxins promotes local bacterial colonization as a result of increased bacterial adhesion, which can occur due to changes in the distribution of receptors on the surface of affected cells with impaired cell morphology. Along with the cells, the functional capacity of the tissues composed of them and the local lymphocytes exposed to toxins also change, which can lead to bacterial invasion and infection [40]. Chronic inflammatory diseases are associated with an increased risk of developing tumors. In the case of bacterial genotoxins, there is an assumption that damaged DNA is the main factor in the acquisition of a malignant phenotype by cells and the onset and/or progression of a tumor, especially in combination with host genetic factors [41]. Figure 1 shows the main events that lead eukaryotic cells to develop a malignant phenotype under the influence of genotoxins.
Colibactin, a nonribosomal polyketide peptide, is a secondary metabolite found in various species of bacteria of the Enterobacteriaceae family, including E. coli, Citrobacter koseri, Klebsiella pneumoniae, and Enterobacter aerogenes, carrying the genomic island of polyketide synthase (pks) on the bacterial chromosome. The origin of pks is not entirely clear, but it has been hypothesized that the clb gene cluster spreads by horizontal transfer (HGT) through ICE-like elements (integrative and conjugative elements, ICEs) and undergoes a stabilization (homing) process upon their chromosomal integration [9,42].
The pks island was originally described in the extraintestinal pathogenic E. coli, ExPEC (Nougayrede et al., 2006) [43]. It has been shown that the presence of the pks island among E. coli is characteristic of the B2 phylogenetic group, which includes most of the ExPEC strains characterized by high virulence, such as expression of the K1 capsule in E. coli causing sepsis and neonatal meningitis [43,44]. The pks gene cluster in K. pneumoniae is identical to E. coli and is also associated with K. pneumoniae hypervirulence [45]. It is noteworthy that the systemic infection of E. coli K1 and K. pneumoniae K1 necessarily includes the invasion of the intestinal mucosa and the stage of intestinal translocation, which is reduced due to impaired colonization of the intestine by these bacteria upon inactivation of individual genes of the pks island-clbA or clbP, in experimental infections of animals [46,47]. The B2 group is associated with the carriage of many pathogenicity factors: adhesins (P- and Type 1 Fimbriae), capsular antigens (K1 and K5), aerobactin, hemolysin, and some pathogenicity islands (PAIs) associated with the persistence of E. coli in the intestinal microbiota. These factors increase the adaptability of bacteria to the normal intestinal environment, and the occurrence of extraintestinal virulence is an accidental by-product of commensalism [48]. The pks island is also often detected in intestinal isolates of E. coli extracted from healthy people, including infants, and is even present in the probiotic strain E. coli Nissle 1917 [49]. Probably, the acquisition of genotoxicity promotes increased colonization of the intestine by pks + bacteria, acting as a factor in the survival of bacteria that create and expand their own niches in the complex microbial ecosystem of the intestine [50,51]. Successful competition for host biotopes of microorganisms is also associated with their antimicrobial activity and intermediate products formed during the synthesis of colibactin, inhibiting the growth of other bacteria—Staphylococcus aureus, Bacillus subtilis—and giving an advantage to bacteria carrying pks genes [50,52].
The pks genomic island of E. coli contains 19 genes (from clbA to clbS), 54 kb in size, encoding the synthesis of colibactin, while the structure of the compound remains unknown due to its high instability. The structure of 13 synthetic derivatives of colibactin was proposed using a synthesis strategy, and it has been shown that the formation of an electrophilic cyclopropane fragment, which can bind to DNA and alkylate adenine of both strands, causing DNA interstrand cross-links, ultimately occurs. This phenomenon has been identified both in HeLa cells and in mouse models [53,54].
The genotoxic activity of colibactin depends on contact with human or animal cells and is not observed when cell lines are treated with pks + E. coli bacterial culture supernatants or their lysates. This contact is facilitated by inflammation when the mucus layer on the surface of the colon is broken, and the contact of the intestine’s surface cells with live pks + E. coli becomes possible [55,56]. Mucus becomes more permeable when mucosal homeostasis is disturbed, including excessive consumption of mucus glycans by microorganisms as an alternative source of nutrients. As a result, bacteria penetrate into the inner layers of mucus, and severe damage can lead to erosion of the colonic mucosa [57]. In addition, many intestinal bacteria interact with mucosal carbohydrates, glycoproteins, and glycolipids to colonize the mucosal surface. Toxin-producing bacteria may have a double advantage in intestinal colonization due to the fact that toxins also attach to host glycan structures. It is likely that the disturbed mucus layer can not only affect the pathogenic and carcinogenic potential of bacteria, but also their ability to penetrate the inner mucus layer and interact with transmembrane mucins covering the apical surface of enterocytes [58].
Subsequent changes in cells, once bacteria are in close contact with them, are associated with DNA double-strand breaks (DSB) accompanied by phosphorylation of replication protein A (pRPA) and histone H2AX (pH2AX), which occur due to replication stress caused by DNA cross-links. The ATM-CHK2 DNA damage checkpoint pathway is activated, and the cell cycle stops at the G2/M stage, resulting in subsequent incomplete DNA repair [50]. By damaging DNA and blocking the cell cycle, the renewal of eukaryotic cells is slowed down, which contributes to the stability and long-term persistence of pks + bacteria in the complex microbial communities of the host intestine [56]. Further events in cells damaged by colibactin can be considered as its side effect, promoting carcinogenesis. Surviving eukaryotic cells are characterized by genomic instability, expressed in chromosomal aberrations, aneuploidy, and an increase in the frequency of gene mutations [9,14]. The role of pks + E. coli in mutagenesis was confirmed by a study conducted on human intestinal organelles derived from primary crypt stem cells. In organelles exposed to pks + E. coli, mutational signatures of SBS-pks were revealed with an increased level of single base substitutions (SBS) in ATA, ATT, and TTT, with the replacement of the middle T and small indel, ID-pks, with single T deletions at homopolymers. In addition, upstream of the SBS-pks sites and upstream of the poly-T site in ID-pks, adenine enrichment was found, which is characteristic of pks signatures and distinguishes them from those of other alkylating agents. Notably, based on the analysis of WGS data from a Dutch collection of solid cancer metastases, SBS-pks and ID-pks signatures were detected mainly in CRC [59].
A dose-dependent effect of pks + E. coli on eukaryotic cells has been shown as follows: low doses induced a transient response to DNA damage followed by resumption of cell division with signs of damaged DNA; high doses of toxigenic bacteria caused irreversible cell cycle arrest and apoptosis [13]. A large number of pks + bacteria causes massive DNA damage and phenotypic cellular senescence, in which the cytotoxic effect of colibactin is observed—megalocytosis, the absence of mitosis [47,54]. Senescent cells begin to secrete pro-inflammatory cytokines, chemokines, proteases, and growth factors into the environment, such as hepatocyte growth factor (HGF), which stimulate the proliferation of uninfected neighboring cells. Therefore, pks + bacteria accelerate the progression of neoplasms, as shown in mouse models of colon tumors [60,61].
Thus, the putative role of colibactin in carcinogenesis may be associated with both the initiation of tumor development and enhancement of tumor growth. In support of this, colibactin-positive bacteria are found with increased frequency in patients with inflammatory bowel disease (IBD), familial adenomatous polyposis, and colorectal cancer (CRC) and are associated with the etiology and pathogenesis of these diseases [62,63]. Accumulating epidemiological evidence indicates the existence of dietary risk factors for colorectal cancer that are metabolized by the gut microbiota and may influence its composition. A high intake of grains or a low intake of vegetables, especially from the cruciferous family, revealed a positive association between pks + E. coli and colorectal neoplasia [19].
Infections of other organs by pks + bacteria are also considered to be an increased risk factor for cancer. Thus, bladder cancer may be preceded by regular urinary tract infections (UTIs) caused by E. coli pks + [11]. It has been shown that in the urine of patients infected with isolates of uropathogenic E. coli (UPEC) pks +, the C14-Asn metabolite, which is formed during the synthesis of colibactin, is detected, and damage by colibactin to the DNA of urothelial cells Krt14 from which a significant part of cancer tissue originates, was experimentally confirmed on rodents. [64].
Cytolethal distending toxin is a genotoxin produced by many Gram-negative bacteria, such as E. coli, Campylobacter spp., Aggregatibacter actinomycetemcomitans, Haemophilus ducreyi, Helicobacter spp., Shigella dysenteriae, Haemophilus spp., Providencia alcalifaciens [10,65,66]. CDT-producing bacteria colonize mucus membranes and skin as commensals or as agents of localized or disseminated infections in mammals [14]. Initially, CDT was detected in pathogenic E. coli upon treatment of Vero, CHO, HeLa, and HEp-2 eukaryotic cells with bacterial culture supernatant, which caused cell elongation and nucleus enlargement 120 h after addition [67].
Morphological changes associated with CDT are a consequence of cell cycle disturbances in infected cells. In most bacteria, the gene cluster encoding CDT subunits is located on the chromosome; in some E. coli strains, it is located on the conjugative plasmid (pVir), which also encodes other virulence determinants [68]. CDT consists of three different protein subunits, A, B, and C, which are encoded by three genes, cdtA, cdtB, and cdtC, respectively. They were first cloned and sequenced from E. coli (O86:H34) (Scott and Kaper, 1994) [69]. The cdtA subunit interacts with receptors on the surface of target cells and delivers the toxin into the cytosol; the cdtC subunit interacts with intracellular transport systems and ensures the transfer of the cdtB toxin into the eukaryotic cell nucleus. Once the cdtB enzyme translocates to the nucleus, it induces DNA single-strand and DSBs and cell cycle arrest at the G1/S or G2/M boundary, depending on the cell type [20,41]. The accumulation of DSB leads to a cellular response to DNA damage involving the ATM (ataxia telangiectasia mutated) system, which activates Chk2, which in turn phosphorylates and inactivates CDC25 phosphatase, resulting in the phosphorylated cyclin B-CDK1 complex preventing mitotic entry. Cell cycle arrest at the G1/S stage by CDT mainly depends on p53: ATM phosphorylates p53 and activates p21, which inhibits cyclin E–CDK2, which in turn blocks entry into the S phase [65]. The arrest of epithelial and endothelial cell lines mainly occurs in the G2/M phase, and the arrest of fibroblast cells in both the G1/S and G2/M phases of the cell cycle, with G1/S arrest observed in primary fibroblast cell lines containing wild-type p53 [10,70]. The subsequent event of CdtB-induced activation of the cellular response may be apoptosis mediated through both p53-dependent and p53-independent pathways. Hematopoietic cells undergo rapid p53-mediated apoptosis; epithelial, endothelial clones, and all p53-deficient cells show resistance to apoptosis, which leads to cellular senescence [71].
Limited damage to host cells stimulates specific cellular responses. Influencing the activity of B- or T-cells, CDT prevents their maturation into effector cells and induces the aging of T-cells in vivo [72]. Microorganisms producing CDT may be the cause of irritable bowel syndrome with diarrhea (IBS-D). Anti-cdtB antibodies cross-react with an intracellular cytoskeletal protein, vinculin, which is a component of cell adhesion and plays a key role in the contractility of nerve cells in the gastrointestinal tract. [73]. Pathogens such as Campylobacter jejuni or E. coli, which trigger the subsequent development of IBS-D, first cause gastroenteritis. The further effect of the toxin on the proliferating epithelial cells in the intestinal crypts leads to the cell cycle arrest in the G2 phase. As a result, cell growth and renewal stop, which facilitates the colonization of the intestine by bacteria. Impaired maturation of epithelial cells of intestinal villi leads to epithelial erosion and loss of absorptive function [39]. Persistent colonization and associated inflammation, invasion, immunosuppression, and DNA damage may influence the onset and development of tumors.
The role of CDT in oncogenesis was confirmed by experiments carried out on GF (germ-free) Apc Min/+ mouse models (He et al., 2019) [74]. It has been shown that genotoxin produced by C. jejuni isolate extracted from human, due to the toxic effect of CdtB, induced DNA damage, contributing to the development of colorectal cancer, while the number and size of tumors in mice decreased when they were infected with bacteria with a mutated CdtB subunit [74]. In the intestinal and liver cell lines, CdtB-dependent regulation of the V-maf musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB) gene has been found [75].
In vitro and in vivo experiments have also demonstrated the possible role of the toxin in tumor progression. In response to infection with bacterial CDT, cytoplasmic invaginations formed by the nucleoplasmic reticulum (NR) were found in target cells. NR structures, being the active sites of mRNA translation, correlated with a high degree of ploidy and were involved in the survival of cells with damaged DNA. Polyploid cells continued to proliferate with NR resorption and returned to normal size, which may contribute not only to the continued growth of the tumor, but also to its resistance to radiation therapy that causes DNA damage [66].
In some cases, the effect of CDT on cancer cells led to their accelerated death associated with the implementation of the strategy of genotoxin-infected cells’ apoptotic death. It turned out that CDT effectively inhibits the growth of radioresistant prostate cancer associated with the loss of DAB2IP expression. DAB2IP is a protein that activates Ras-GTPase and regulates proliferation and apoptosis by inactivating PI3K-Akt. The toxin enhances the effect of IR as a result of prolonged cell cycle arrest at G2/M followed by apoptosis induction [76].
5. Specific Microbial Metabolites
Bacterial populations in the human body and specific metabolites associated with them support physiological functions in its various biotopes. Depending on the genetic and epigenetic background of the host, an imbalance between the protective and pathological functions of microbial metabolites can lead to the occurrence of diseases, including cancer [15,77]. Figure 2 shows the main events that lead eukaryotic cells to develop a malignant phenotype when exposed to bacterial metabolites.
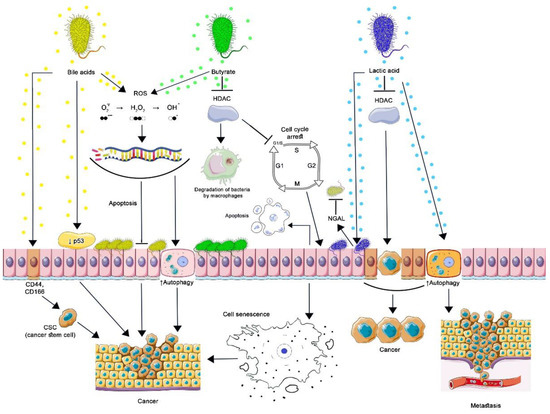
Figure 2.
Contribution of commensal bacteria microbial metabolites to carcinogenesis. By stimulating the proteasomal degradation of p53 and inducing cancer stem cells (CSC), secondary BAs lead to malignant transformation of cells. Oxidative damage to DNA by secondary BA and butyrate is associated with the formation of ROS, which cannot be restored in genetically modified cells, which leads to the formation of resistance to apoptosis, increased autophagy, and cancer development. Butyrate inhibits HDAC in normal and cancer cells. The action of butyrate upon HDAC3 of intestinal macrophages reduces the activity of the mTOR protein, as a result of which autophagy in macrophages is enhanced. This is how butyrate-producing bacteria maintain the optimal level of their population. By inhibiting HDAC, butyrate induces the expression of p21, which arrests the cell cycle at the G1-S stage: cells can undergo apoptosis or acquire a senescence-associated secretory phenotype leading to the development or progression of cancer. By inhibiting the production of cyclic adenosine monophosphate (cAMP), lactic acid activates autophagy in vaginal epithelial cells, protecting them from intracellular microorganisms. Protective autophagy in tumor cells prevents apoptosis, promoting tumor metastasis. By blocking HDAC, lactic acid increases the production of NGAL by epithelial cells, which inhibits the growth of other microorganisms, inducing selective transcription of genes in malignantly transformed cells, which contributes to their survival and stimulates oncogenesis. (Figure 2 was created using images from Servier Medical Art https://smart.servier.com/ 26 June 2022).
Butyrate is an organic metabolite of the colon microbiota that plays a key role in maintaining homeostasis and regulating intestinal barrier function. The main producers of butyrate in the human intestine are obligate anaerobes belonging to phylogenetically diverse groups of bacteria: Faecalibacterium prausnitzii and Roseburia spp., which belong to Ruminococcaceae (Clostridial cluster IV) and Lachnospiraceae (Clostridial cluster XIVa) families of the phylum Firmicutes, respectively [78,79,80].
Energy metabolism and microbial ecology are common to groups of butyrate-producing bacteria; therefore, to maintain their populations, they compete with carbohydrate-consuming Bacteroides spp. and have an advantage at a lower pH (5.5) of the proximal colon. This moderately acidic pH level occurs when there is a high level of bacterial fermentation of carbohydrates in the colon, which are not digested in the upper intestine [81]. Butyrate has a direct antimicrobial effect on bacteria, damaging their cell membranes and acidifying the cytosol, as well as increasing the production of host defense peptides (HDP) by intestinal epithelial cells: RegIIIγ, β-defensins, cathelicidin LL-37 [82,83]. Acting as a differentiation factor, butyrate induces metabolic and transcriptional changes in monocyte-derived macrophages, enhancing their antimicrobial functions. These changes are associated with the action of butyrate as an inhibitor of histone deacetylases (HDAC)—enzymes that remove histone acetyl groups. HDAC inhibitors maintain a high level of histone acetylation at promoter loci, providing an epigenetic mechanism that regulates their expression. As a result of butyrate’s effect on HDAC3 of intestinal macrophages, they increase the expression of calprotectin, a protein that enhances the destruction of bacteria through zinc ions (Zn2+) and manganese (Mn2+) sequestration, decreases the activity of the mTOR protein, which stops the suppression of autophagy, and increases the production of LC3, a protein involved in the formation of autophagosomes [83,84]. Butyrate triggers a phagocytic antimicrobial defense program in the absence of an increased inflammatory cytokine response; in addition, butyrate reduces the production of pro-inflammatory mediators such as NO, IL-6, and IL-12. Thus, butyrate-producing bacteria, by regulating HDAC and avoiding the destructive effects of inflammation, contribute to maintaining the optimal level of their population in the intestine [84,85].
Butyrate plays an exceptional role in intestinal homeostasis and has an anticarcinogenic effect on cancer cells of various genesis, which is well described in numerous literature sources. But do the underlying mechanisms of the antitumor effect of butyrate lead to the expected result in all cases? We tried to figure that out.
Butyrate, as an HDAC inhibitor, has antiproliferative properties against tumor cells. Normal colonocytes use butyrate as the main source of energy, while cancerous colonocytes use glucose during glycolysis (Warburg effect). Therefore, butyrate can accumulate in the nucleus and function as an HDAC inhibitor, increasing the expression of target genes and preventing the proliferation of cancerous colonocytes [86]. For instance, inhibition of HDAC in tumor cell lines induces p21 expression. This inhibitor of cyclin E (cyclin-dependent kinase 2, Cdk2) activity stops the cell cycle at the G1-S stage, while non-proliferating cells differentiate or undergo apoptosis, which can be interpreted as effects that limit the occurrence and progression of cancer [87]. Although irreversible cell cycle arrest initially acts as a defense mechanism against carcinogenesis, it is believed that this leads to an accumulation of cells with a senescence-associated secretory phenotype (SASP), which can lead to tumor progression. CRC-associated butyrate producers Porphyromonas gingivalis and Porphyromonas asaccharolytica have been found to induce a senescence-associated secretory phenotype (SASP) in cultured intestinal epithelial cells and organelles as well as accelerate colorectal tumor development in mice. SASP cells contribute to the accumulation of immune cells in their environment and maintain chronic inflammation and disruption of the epithelial cell barrier integrity due to impaired tight junctions between them, which can contribute to carcinogenesis [88].
Differences in the host genotype can also lead to a multidirectional effect of butyrate on colonic epithelial cells, which has been demonstrated in experiments on mouse models. Butyrate stimulated the formation of colorectal polyps in mice with the APC Min/+ genotype, MSH2 −/−. Mice with multiple intestinal neoplasia, APC Min/+) represent an animal model of human adenomatous polyposis. Furthermore, these mice were deficient in the DNA repair gene, 2 MutS homolog (MSH2 -/-), which is involved in correcting errors that occur during DNA replication. Presumably, MSH2 deficiency intensifies genetic changes in the tumor, which can lead to increased cell proliferation in response to the action of butyrate [89,90].
Butyrate is actively produced by commensal bacteria and absorbed by colonocytes from the colonic lumen, then it undergoes β-oxidation until acetyl-CoA forms in mitochondria, maintaining energy homeostasis and preventing cell autophagy [78,79]. On the other hand, the mitochondrial oxidative metabolism of butyrate is the main mechanism that increases the amount of ROS in intestinal epithelial cells. During normal metabolism or inflammation, ROS-induced DNA damage in colonocytes is repaired by the BER (base excision repair) and MMR (mismatch repair) systems. However, in the absence of the MMR system in tissues, as shown in mouse models, butyrate leads to the accumulation of ROS and oxidative DNA damage. In the case of both Lynch syndrome and the sporadic form of CRC, MMR genes are defective and cannot repair damage caused by ROS, which leads to the accumulation of oxidative stress product, 8-oxo-dG, in the body [80].
Also interesting are the new data on the development of resistance to chemotherapy of CRC cells, which occurs from the resistance of CRC cells to butyrate. CRC patients responding to chemotherapy have a high abundance of butyrate-producing bacteria. Butyrate resistance is formed by the efflux mechanism as a result of an increase in the expression level of efflux genes. At the same time, this mechanism contributes to the formation of cross-resistance of HCT-116 cells to metformin and oxaliplatin and PMF-K014 cells to 5-fluorouracil [91].
Lactic acid. A significant part of the microbiota of various human loci is made up of bacteria that produce lactic acid, the main nutrient substrate for which is epithelial cell glycogen. Species L. crispatus, L. iners, L. gasseri, and L. jensenii produce especially large amounts of lactic acid in the vagina [92]. Lactobacilli produce lactic acid, which lowers the pH of the extracellular medium, creating favorable conditions for their own survival and preventing the adhesion and growth of other microorganisms [93]. The protonated form of lactic acid, which is permeable to membranes without the use of monocarboxylate transporters and the GPR81 receptor, has antimicrobial and immunomodulatory activity. Lactic acid, penetrating into bacterial cells, acidifies the cytosol and increases the permeability of the cell membrane, disrupting its intracellular functions, which leads to the death of bacteria [94].
The antimicrobial activity of lactic acid is aimed at potentially pathogenic microorganisms and different infectious agents, while lactic acid induces immune tolerance, reducing the activity of cytotoxic T lymphocytes and natural killer cells, inhibiting the release of pro-inflammatory cytokines from epithelial cells and the maturation of dendritic cells. Similarly, lactic acid, formed by tumor cells as a result of the predominance of anaerobic metabolism, creates a favorable tumor microenvironment by blocking the transcription of the interferon-γ gene, contributing to the weakening of antitumor immunity, inducing HIF-1α to enhance the proliferation of cancer cells, inducing the synthesis of hyaluronan by tumor cells to destroy extracellular matrix, and increasing their metastatic potential [95].
Lactic acid inhibits the production of cyclic adenosine monophosphate (cAMP), activating autophagy in vaginal epithelial cells, thereby protecting them from oxidative stress resulting from the intracellular accumulation of defective mitochondria, as well as from intracellular microorganisms [96,97]. Despite the fact that autophagy is a mechanism that prevents the development of cancer, including the death of transformed cells in the early stages of the disease, protective autophagy prevents apoptosis in tumor cells, promoting tumor metastasis and the development of resistance to radiation and chemotherapeutic agents, for example, the progression of endometrial cancer is associated with increased autophagy [95,98].
Lactic acid is an epigenetic regulator of gene activity, blocks HDAC, and enhances the transcription of genes responsible for components of the innate immune system, such as neutrophil gelatinase-associated lipocalin (NGAL). NGAL is present in neutrophils and many other tissues. Produced by epithelial cells in the presence of lactobacilli, NGAL suppresses bacterial siderophores, preventing the acquisition of iron by microorganisms, thereby inhibiting their growth, except for lactobacilli. D-lactic acid—a specific bacterial metabolite—is a more potent HDAC inhibitor compared to the L-isomer [96,99]. HDACs control the repression of non-expressed genes and also have a repressive effect on actively expressed genes. As a result, there is a weakening of transcription in active genes. Lactate, as an HDAC inhibitor, is able to preserve the transcription of active genes in order to avoid temporary glycogen depletion caused by increased glycolysis and maintains cell viability, which is also important for tumor cells. As experimental data show, inhibition of HDAC by lactic acid occurs at concentrations found in nature [100].
It has been shown that lactic acid produced by the bacteria Lactobacillus, Lactococcus, Leuconostoc, Pediococcus, Streptococcus, and Peptostreptococcus is involved in the development of oral squamous cell carcinoma by lowering the pH of the environment, enhancing gene transcription and DNA repair by inhibiting HDAC. It is likely that HDAC inhibition of actively expressed genes contributed to the survival of tumor cells. It is also noteworthy that the number of Lactobacillus has increased in accordance with the TNM stage increase in cancer patients after radiation and chemotherapy. Thus, by inducing selective gene transcription as an HDAC inhibitor, lactic acid can promote the survival of malignantly transformed cells and stimulate oncogenesis [101,102].
Secondary bile acids. Primary bile acids (BA)—cholic (CA) and chenodeoxycholic (CDCA) acids synthesized by liver cells—go through the enterohepatic circulation in the human body; one of the stages of which is the formation of secondary bile acids. Deoxycholic acid (DCA) and lithocholic acid (LCA) are formed, respectively, from CA and CDCA as a result of bacterial enzymatic dehydroxylation reactions in the large intestine [103]. The main producers of DCA and LCA are intestinal commensals of the Clostridium genus, which are part of cluster XVI (Clostridium cluster XVI): C. scindens, C. hylemonae, and cluster XI (Clostridium cluster XI): C. sordelli, C. bifermentans, C. hiranonis (Peptacetobacter hiranonis) [104,105].
The deconjugated primary bile salts cholate and chenodeoxycholate undergo reductive 7-dehydroxylation and are used by bacteria as electron acceptors, forming NAD+. Electron utilization by 7-dehydroxylating bacteria as a result of the conversion of primary FAs into secondary FAs provides this group of bacteria with a selective advantage in the process of colonization of the host’s intestine [106].
Secondary Bas—more hydrophobic and toxic than primary Bas—inhibits the growth of many intestinal bacteria by damaging their membranes, acting as detergents. Thus, the minimum inhibitory concentration of DCA against Lactobacillus and Bifidobacterium species is ten times lower than that of CA [107]. To reduce the toxicity of bile acids, intestinal microorganisms oxidize the hydroxyl groups of both primary and secondary BAs with the help of microbial HSDH (hydroxysteroid dehydrogenase) enzymes. However, some 7α-dehydroxylating bacteria have developed additional mechanisms that maintain antagonistic interactions with competing bacterial species by repeated reduction of the oxidized form of LCA (12-oxoLCA) [106,108].
In addition, the ability to produce antibacterial compounds helps 7α/β-dehydroxylating bacteria occupy their ecological niche in the large intestine. C. scindens has been shown to secrete tryptophan-derived antibiotics that inhibit the growth of C. difficile. The secretion of antibacterial compounds is enhanced by DCA and LCA, especially DCA, and these compounds themselves increase the toxicity of bile acids and the effectiveness of the suppression of intestinal bacteria [109]. In addition, secondary bile acids negatively affect spore germination, growth kinetics, and toxin activity of clinically significant strains of C. difficile, preventing the development of diseases associated with this bacterium [110]. Bile acids can also have an indirect effect on pathogens of intestinal infections by changing the composition of the microbiota. In experiments to prevent the colonization of chickens by the causative agent of campylobacteriosis C. jejuni, DCA was used, which had no inhibitory effect on C. jejuni in vitro. As a result, DCA-treated birds had a decreased amount of Firmicutes, an increased amount of Bacteroidetes, and decreased C. jejuni colonization [105].
Protecting the host from pathogens, DCA and LCA are toxic not only to the microbiota, but also to the host. Most LCA is esterified with sulfate in the human liver and excreted in the bile [111]. Due to the lack of a metabolic pathway to remove DCA, it accumulates in the bile acid pool. The amount of DCA in the pool depends on the rate of formation and absorption of DCA through the colon, the transit time, and the pH of the colon. With an increase in pH, the solubility of DCA increases, and therefore it is easily absorbed in the colon and enters the bloodstream. With a decrease in pH, an excess amount of deoxycholic acid can bind to dietary fiber and be excreted from the body [112].
The toxicity of secondary bile acids is also associated with carcinogenesis. They act as tumor promoters by modulating intracellular signaling and gene expression. DCA reduces p53 protein accumulation by stimulating proteasomal degradation. Being in a suppressed state, p53 and its response to DNA damaging agents such as ionizing radiation can manifest itself as the inability of cells to eliminate tumor transformation [113]. Interactions between DNA in Pirc rats with a germline mutation in the Apc—a tumor suppressor gene (TSG)—and the gut microbiota during carcinogenesis have been researched. The appearance of adenomas in the Pirc mucosa was associated with increased oxidative DNA damage, which was likely related to gut microbiota metabolism. Notably, the Pirc colon was enriched in 7-dehydroxylated bacteria Clostridium cluster XI [114]. In addition to oxidative damage to DNA by secondary BA associated with the formation of ROS and nitrogen, the impact of increasing concentrations of DCA on colonic epithelial cell cultures led to the formation of apoptosis resistance, and in experiments with animals, an increase in the expression of the anti-apoptotic protein Bcl-xL in the area adjacent to adenocarcinomas [115]. Secondary BAs are able to induce cancer stem cells (CSC) in colon epithelial cells. Given the key role of the Wnt/β-catenin signaling pathway in the regulation of colonic CSC proliferation, it has been shown that under the influence of DCA and LCA in HCoEpiC cells, the level of CSC markers (CD44 and CD166) has increased, as well as the expression of M3R, β-catenin, c-Myc, indicating cell transformation into CSCs by modulating M3R and Wnt/β-catenin signaling [116]. Microbial metabolites may be involved in the epigenetic regulation of cellular processes by influencing miRNA expression. Deoxycholic acid promotes the development of CRC by downregulating miR-199a-5p, which acts as an anti-oncogenic miRNA, inhibiting the proliferation, migration, and invasion of colorectal cancer cells in vitro and in vivo indirectly through CAC1 protein. This protein interacts with CDK2, activating the cell cycle, and the loss of CAC1 properties leads to its arrest in the G1/S phase [117,118].
6. Conclusions
In the course of co-evolution with macroorganisms, bacteria have developed effective mechanisms of interaction and adapted to their specific niches. The role of many commensals is determined by traditional ideas about their useful functions and maintaining homeostasis of the body’s biotopes, mainly through the synthesis of biologically active compounds. However, the survival of microorganisms and their successful colonization of various organs and systems of the host may be associated with the development of pathological effects, including the development and progression of tumors.
Microorganisms that synthesize cyclomodulins do little harm to the cells of the body in order to colonize it for a long time. Under unfavorable external conditions and a disturbed internal environment of the host, the effect of toxins on the eukaryotic cells of the body leads to DNA damage, disruption of the cell cycle, and mutational events that contribute to carcinogenesis.
Bacteria producing butyrate, lactic acid, and secondary bile acids limit exogenous microbial colonization either by the direct effect of these compounds on competing microorganisms or through host defense mechanisms or antimicrobial agents whose activity is associated with these metabolites. The same metabolites, in response to environmental factors, altered genetic background of the macroorganism, phenotypic, physiological, and biochemical diversity of its cells, can participate in carcinogenesis. The consequential imbalance between the state of the macroorganism and the factors of bacteria adaptation to it—cyclomodulins and some other microbial metabolites—can be the cause of the onset and development of malignant transformations in the host organism.
Author Contributions
Conceptualization, methodology, writing—review, N.N.M.; formal analysis, editing, and mentoring, N.N.M., E.F.S.; visualization, O.N.S.; investigation, editing, and project administration, N.N.M. and V.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Russian Science Foundation (grant no. 22-25-00353).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Special thanks to Tiumin V. Boris for providing the English translation of this article and also for his tremendous help in editing and creating the figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Silva-García, O.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M. Wnt/β-catenin signaling as a molecular target by pathogenic bacteria. Front. Immunol. 2019, 10, 2135. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; DePaolo, R.W. “Driver-passenger” bacteria and their metabolites in the pathogenesis of colorectal cancer. Gut Microbes 2021, 13, 1941710. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef]
- El-Aouar Filho, R.A.; Nicolas, A.; De Paula Castro, T.L.; Deplanche, M.; De Carvalho Azevedo, V.A.; Goossens, P.L.; Taieb, F.; Lina, G.; Le Loir, Y.; Berkova, N. Heterogeneous Family of Cyclomodulins: Smart Weapons That Allow Bacteria to Hijack the Eukaryotic Cell Cycle and Promote Infections. Front. Cell Infect. Microbiol. 2017, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef]
- Rosadi, F.; Fiorentini, C.; Fabbri, A. Bacterial protein toxins in human cancers. Pathog. Dis. 2016, 74, ftv105. [Google Scholar] [CrossRef]
- Mezerová, K.; Starý, L.; Zbořil, P.; Klementa, I.; Stašek, M.; Špička, P.; Skalický, P.; Raclavský, V. Cyclomodulins and Hemolysis in E. coli as Potential Low-Cost Non-Invasive Biomarkers for Colorectal Cancer Screening. Life 2021, 11, 1165. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Nougayrède, J.P. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The biology of the cytolethal distending toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Aung, K.M.; Uhlin, B.E.; Wai, S.N. Reversible senescence of human colon cancer cells after blockage of mitosis/cytokinesis caused by the CNF1 cyclomodulin from Escherichia coli. Sci. Rep. 2018, 8, 17780. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrède, J.P.; Watrin, C.; Jubelin, G.; Oswald, E.; Taieb, F. Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell Microbiol. 2008, 10, 2496–2508. [Google Scholar] [CrossRef]
- Barrett, M.; Hand, C.K.; Shanahan, F.; Murphy, T.; O’Toole, P.W. Mutagenesis by microbe: The role of the microbiota in shaping the cancer genome. Trends Cancer 2020, 6, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Duijster, J.W.; Franz, E.; Neefjes, J.; Mughini-Gras, L. Bacterial and Parasitic Pathogens as Risk Factors for Cancers in the Gastrointestinal Tract: A Review of Current Epidemiological Knowledge. Front. Microbiol. 2021, 8, 790256. [Google Scholar] [CrossRef]
- de Savornin Lohman, E.; Duijster, J.; Groot Koerkamp, B.; van der Post, R.; Franz, E.; Mughini Gras, L.; de Reuver, P. Severe Salmonella spp. or Campylobacter spp. Infection and the Risk of Biliary Tract Cancer: A Population-Based Study. Cancers 2020, 12, 3348. [Google Scholar] [CrossRef]
- Iwasaki, M.; Kanehara, R.; Yamaji, T.; Katagiri, R.; Mutoh, M.; Tsunematsu, Y.; Sato, M.; Watanabe, K.; Hosomi, K.; Kakugawa, Y.; et al. Association of Escherichia coli containing polyketide synthase in the gut microbiota with colorectal neoplasia in Japan. Cancer Sci. 2022, 113, 277–286. [Google Scholar] [CrossRef]
- Grasso, F.; Frisan, T. Bacterial genotoxins: Merging the DNA damage response into infection biology. Biomolecules 2015, 5, 1762–1782. [Google Scholar] [CrossRef]
- Chaoprasid, P.; Lukat, P.; Mühlen, S.; Heidler, T.; Gazdag, E.M.; Dong, S.; Blankenfeldt, W. Crystal structure of bacterial cytotoxic necrotizing factor CNFY reveals molecular building blocks for intoxication. EMBO J. 2021, 40, e105202. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Mettouchi, A.; Wilson, B.A.; Lemichez, E. CNF1-like deamidase domains: Common Lego bricks among cancer-promoting immunomodulatory bacterial virulence factors. Pathog. Dis. 2018, 76, fty045. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The cytotoxic necrotizing factor 1 from E. coli: A janus toxin playing with cancer regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef]
- Garcia, T.A.; Ventura, C.L.; Smith, M.A.; Merrell, D.S.; O’Brien, A.D. Cytotoxic necrotizing factor 1 and hemolysin from uropathogenic Escherichia coli elicit different host responses in the murine bladder. Infect. Immun. 2013, 81, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Carlini, F.; Maroccia, Z.; Fiorentini, C.; Travaglione, S.; Fabbri, A. Effects of the Escherichia coli Bacterial Toxin Cytotoxic Necrotizing Factor 1 on Different Human and Animal Cells: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 12610. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Rosadi, F.; Ballan, G.; Maroccia, Z.; Giambenedetti, M.; Fiorentini, C. The Escherichia coli protein toxin cytotoxic necrotizing factor 1 induces epithelial mesenchymal transition. Cell. Microbiol. 2020, 22, e13138. [Google Scholar] [CrossRef]
- De Rycke, J.E.A.N.; Mazars, P.; Nougayrede, J.P.; Tasca, C.; Boury, M.; Herault, F.; Oswald, E. Mitotic block and delayed lethality in HeLa epithelial cells exposed to Escherichia coli BM2-1 producing cytotoxic necrotizing factor type 1. Infect. Immun. 1996, 64, 1694–1705. [Google Scholar] [CrossRef]
- Tantillo, E.; Colistra, A.; Vannini, E.; Cerri, C.; Pancrazi, L.; Baroncelli, L.; Caleo, M. Bacterial toxins and targeted brain therapy: New insights from cytotoxic necrotizing factor 1 (CNF1). Int. J. Mol. Sci. 2018, 19, 1632. [Google Scholar] [CrossRef]
- Vannini, E.; Mori, E.; Tantillo, E.; Schmidt, G.; Caleo, M.; Costa, M. CTX-CNF1 Recombinant Protein Selectively Targets Glioma Cells In Vivo. Toxins 2021, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Chavez, C.V.; Taieb, F.; Banfield, M.J.; Samba-Louaka, A.; Nobe, R.; Nougayrède, J.P.; Zumbihl, R.; Givaudan, A.; Escoubas, J.M.; et al. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS ONE 2009, 4, e4855. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrède, J.P.; Watrin, C.; Oswald, E.; Taieb, F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect. Immun. 2009, 77, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Loukiadis, E.; Nobe, R.; Herold, S.; Tramuta, C.; Ogura, Y.; Ooka, T.; Morabito, S.; Kérourédan, M.; Brugère, H.; Schmidt, H.; et al. Distribution, functional expression, and genetic organization of Cif, a phage-encoded type III-secreted effector from enteropathogenic and enterohemorrhagic Escherichia coli. J. Bacteriol. 2008, 190, 275–285. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrède, J.P.; Oswald, E. Cycle inhibiting factors (cifs): Cyclomodulins that usurp the ubiquitin-dependent degradation pathway of host cells. Toxins 2011, 3, 356–368. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of Ubiquitin/NEDD8 by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef]
- Piciocchi, A.; Germinario, E.A.P.; Garcia Etxebarria, K.; Rossi, S.; Sanchez-Mete, L.; Porowska, B.; Stigliano, V.; Trentino, P.; Oddi, A.; Accarpio, F.; et al. Association of Polygenic Risk Score and Bacterial Toxins at Screening Colonoscopy with Colorectal Cancer Progression: A Multicenter Case-Control Study. Toxins 2021, 13, 569. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12, 63. [Google Scholar] [CrossRef]
- Pokkunuri, V.; Pimentel, M.; Morales, W.; Jee, S.R.; Alpern, J.; Weitsman, S.; Chang, C. Role of cytolethal distending toxin in altered stool form and bowel phenotypes in a rat model of post-infectious irritable bowel syndrome. Neurogastroenterol. Motil. 2012, 18, 434. [Google Scholar] [CrossRef]
- De Rycke, J.; Oswald, E. Cytolethal distending toxin (CDT): A bacterial weapon to control host cell proliferation? FEMS Microbiol. Lett. 2001, 203, 141–148. [Google Scholar] [CrossRef]
- Lai, Y.R.; Chang, Y.F.; Ma, J.; Chiu, C.H.; Kuo, M.L.; Lai, C.H. From DNA Damage to Cancer Progression: Potential Effects of Cytolethal Distending Toxin. Front. Immunol. 2021, 12, 760451. [Google Scholar] [CrossRef] [PubMed]
- Wami, H.; Wallenstein, A.; Sauer, D.; Stoll, M.; von Bünau, R.; Oswald, E.; Dobrindt, U. Insights into evolution and coexistence of the colibactin-and yersiniabactin secondary metabolite determinants in enterobacterial populations. Microb. Genom. 2021, 7, 000577. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 2008, 46, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Korena, K.; Karpiskova, R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development-systematic review. Toxicon 2021, 197, 126–135. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Martin, P.; Cloup, E.; Stabler, R.A.; Oswald, E.; Taylor, P.W. The genotoxin colibactin is a determinant of virulence in Escherichia coli K1 experimental neonatal systemic infection. Infect. Immun. 2015, 83, 3704–3711. [Google Scholar] [CrossRef]
- Lu, M.C.; Chen, Y.T.; Chiang, M.K.; Wang, Y.C.; Hsiao, P.Y.; Huang, Y.J.; Lai, Y.C. Colibactin contributes to the hypervirulence of pks+ K1 CC23 Klebsiella pneumoniae in mouse meningitis infections. Front. Cell. Infect. Microbiol. 2017, 7, 103. [Google Scholar] [CrossRef]
- Le Gall, T.; Clermont, O.; Gouriou, S.; Picard, B.; Nassif, X.; Denamur, E.; Tenaillon, O. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol. Biol. Evol. 2007, 24, 2373–2384. [Google Scholar] [CrossRef]
- Nougayrède, J.P.; Chagneau, C.V.; Motta, J.P.; Bossuet-Greif, N.; Belloy, M.; Taieb, F.; Gratadoux, J.J.; Thomas, M.; Langella, P.; Oswald, E.A. Toxic Friend: Genotoxic and Mutagenic Activity of the Probiotic Strain Escherichia coli Nissle 1917. mSphere 2021, 6, e0062421. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More than a new bacterial toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef]
- Tronnet, S.; Floch, P.; Lucarelli, L.; Gaillard, D.; Martin, P.; Serino, M.; Oswald, E. The genotoxin colibactin shapes gut microbiota in mice. mSphere 2020, 5, e00589-20. [Google Scholar] [CrossRef] [PubMed]
- Faïs, T.; Cougnoux, A.; Dalmasso, G.; Laurent, F.; Delmas, J.; Bonnet, R. Antibiotic activity of Escherichia coli against multiresistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 6986–6988. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Balskus, E.P. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Kurnick, S.A.; Mannion, A.J.; Feng, Y.; Madden, C.M.; Chamberlain, P.; Fox, J.G. Genotoxic Escherichia coli strains encoding Colibactin, Cytolethal Distending Toxin, and Cytotoxic necrotizing factor in laboratory rats. Comp. Med. 2019, 69, 103–113. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Reuter, C.; Alzheimer, M.; Walles, H.; Oelschlaeger, T.A. An adherent mucus layer attenuates the genotoxic effect of colibactin. Cell. Microbiol. 2018, 20, e12812. [Google Scholar] [CrossRef] [PubMed]
- Melhem, H.; Regan-Komito, D.; Niess, J.H. Mucins Dynamics in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2021, 22, 13642. [Google Scholar] [CrossRef] [PubMed]
- Josenhans, C.; Müthing, J.; Elling, L.; Bartfeld, S.; Schmidt, H. How bacterial pathogens of the gastrointestinal tract use the mucosal glyco-code to harness mucus and microbiota: New ways to study an ancient bag of tricks. Int. J. Med. Microbiol. 2020, 310, 151392. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Gagnière, V.; Bonnin, A.S.; Jarrousse, E.; Cardamone, A.; Agus, N.; Uhrhammer, P.; Sauvanet, P.; Déchelotte, N.; Barnich, R.; Bonnet, D.; et al. Bonnet Interactions between microsatellite instability and human gut colonization by Escherichia coli in colorectal cancer. Clin. Sci. 2017, 131, 471–485. [Google Scholar] [CrossRef]
- Taieb, F.; Petit, C.; Nougayrede, J.P.; Oswald, E. The Enterobacterial Genotoxins: Cytolethal Distending Toxin and Colibactin. EcoSal Plus 2016, 7(1), 21. [Google Scholar] [CrossRef] [PubMed]
- Iyadorai, T.; Mariappan, V.; Vellasamy, K.M.; Wanyiri, J.W.; Roslani, A.C.; Lee, G.K.; Vadivelu, J. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS ONE 2020, 15, e0228217. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Darfeuille-Michaud, A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, S.H.; Hanum, N.; Grotenhuis, A.J.; Castano-Vinyals, G.; Van Der Heijden, A.G.; Aben, K.K.; Kiemeney, L.A. Recurrent urinary tract infection and risk of bladder cancer in the Nijmegen bladder cancer study. Br. J. Cancer 2015, 112, 594–600. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Serres, A.; Bonnet, R.; Dalmasso, G. Impact of CDT Toxin on Human Diseases. Toxins 2016, 8, 220. [Google Scholar] [CrossRef]
- Azzi-Martin, L.; He, W.; Péré-Védrenne, C.; Korolik, V.; Alix, C.; Prochazkova-Carlotti, M.; Morel, J.L.; Le Roux-Goglin, E.; Lehours, P.; Djavaheri-Mergny, M.; et al. Cytolethal distending toxin induces the formation of transient messenger-rich ribonucleoprotein nuclear invaginations in surviving cells. PLoS Pathog. 2019, 15, e1007921. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. Response of Chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23. [Google Scholar] [CrossRef]
- Javadi, M.; Bouzari, S.; Oloomi, M. Horizontal gene transfer and the diversity of Escherichia coli. In Escherichia coli—Recent Advances on Physiology, Pathogenesis and Biotechnological Applications; Amidou, S., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157 Pt 7, 1851–1875. [Google Scholar] [CrossRef]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.P.; Namini, M.R.J.; Hansen, M.B.; Martin, O.C.B.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.; Park, S.C.; Morales, W.; Marsh, E.; Lembo, A.; Kim, J.H.; Pimentel, M. Assessment of anti-vinculin and anti-cytolethal distending toxin B antibodies in subtypes of irritable bowel syndrome. Dig. Dis. Sci. 2017, 62, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Péré-Védrenne, C.; He, W.; Azzi-Martin, L.; Prouzet-Mauléon, V.; Buissonnière, A.; Cardinaud, B.; Ménard, A. The nuclear remodeling induced by Helicobacter cytolethal distending toxin involves MAFB oncoprotein. Toxins 2020, 12, 174. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, C.S.; Liu, H.H.; Tsai, Y.S.; Hsu, F.M.; Yu, Y.L.; Lai, C.K.; Gandee, L.; Pong, R.C.; Hsu, H.W.; et al. Sensitization of radio-resistant prostate cancer cells with a unique cytolethal distending toxin. Oncotarget 2014, 5, 5523–5534. [Google Scholar] [CrossRef]
- Tarashi, S.; Siadat, S.D.; Ahmadi Badi, S.; Zali, M.; Biassoni, R.; Ponzoni, M.; Moshiri, A. Gut Bacteria and their Metabolites: Which One Is the Defendant for Colorectal Cancer? Microorganisms 2019, 7, 561. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Hermansen, K. Impact of diet-modulated butyrate production on intestinal barrier function and inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; Van de Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Schauber, J.; Svanholm, C.; Termen, S.; Iffland, K.; Menzel, T.; Scheppach, W.; Melcher, R.; Agerberth, B.; Lührs, H.; Gudmundsson, G. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: Relevance of signalling pathways. Gut 2003, 52, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Powrie, F. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 2019, 50, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell. 2012, 48, 612–626. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Nittayaboon, K.; Leetanaporn, K.; Sangkhathat, S.; Roytrakul, S.; Navakanitworakul, R. Characterization of Butyrate-Resistant Colorectal Cancer Cell Lines and the Cytotoxicity of Anticancer Drugs against These Cells. BioMed Res. Int. 2022. [Google Scholar] [CrossRef]
- Tester, R.; Al-Ghazzewi, F.H. Intrinsic and extrinsic carbohydrates in the vagina: A short review on vaginal glycogen. Int. J. Biol. Macromol. 2018, 112, 203–206. [Google Scholar] [CrossRef]
- Masood, M.I.; Qadir, M.I.; Shirazi, J.H.; Khan, I.U. Beneficial effects of lactic acid bacteria on human beings. Crit. Rev. Microbiol. 2011, 37, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Aldunate, M.; Bradshaw, C.S.; Cone, R.A. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 2017, 168, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S. Lactic acid alleviates stress: Good for female genital tract homeostasis, bad for protection against malignancy. Cell Stress Chaperones 2018, 23, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG 2017, 124, 606–611. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Autophagy: Supporting cellular and organismal homeostasis by self-eating. Int. J. Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Anwer, F. Role and mechanism of autophagyregulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef]
- Nasioudis, D.; Witkin, S.S. Neutrophil gelatinase-associated lipocalin and innate immune responses to bacterial infections. Med. Microbiol. Immunol. 2015, 204, 471–479. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acid Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- La Rosa, G.R.M.; Gattuso, G.; Pedullà, E.; Rapisarda, E.; Nicolosi, D.; Salmeri, M. Association of oral dysbiosis with oral cancer development. Oncol. Lett. 2020, 19, 3045–3058. [Google Scholar] [CrossRef]
- Sami, A.; Elimairi, I.; Stanton, C.; Ross, R.P.; Ryan, C.A. The Role of the Microbiome in Oral Squamous Cell Carcinoma with Insight into the Microbiome–Treatment Axis. Int. J. Mol. Sci. 2020, 21, 8061. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Fujisawa, T. Analysis of Clostridium cluster XI bacteria in human feces. Biosci. Microbiota Food Health 2019, 38, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Alrubaye, B.; Abraha, M.; Almansour, A.; Bansal, M.; Wang, H.; Kwon, Y.M.; Sun, X. Microbial metabolite deoxycholic acid shapes microbiota against Campylobacter jejuni chicken colonization. PLoS ONE 2019, 14, e0214705. [Google Scholar] [CrossRef] [PubMed]
- Doden, H.L.; Ridlon, J.M. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef]
- Kurdi, P.; Kawanishi, K.; Mizutani, K.; Yokota, A. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J. Bacteriol. 2006, 188, 1979–1986. [Google Scholar] [CrossRef]
- Doden, H.; Sallam, L.A.; Devendran, S.; Ly, L.; Doden, G.; Daniel, S.L.; Ridlon, J.M. Metabolism of oxo-bile acids and characterization of recombinant 12α-hydroxysteroid dehydrogenases from bile acid 7α-dehydroxylating human gut bacteria. Appl. Environ. Microbiol. 2018, 84, e00235-18. [Google Scholar] [CrossRef]
- Kang, J.D.; Myers, C.J.; Harris, S.C.; Kakiyama, G.; Lee, I.K.; Yun, B.S.; Hylemon, P.B. Bile acid 7α-dehydroxylating gut bacteria secrete antibiotics that inhibit Clostridium difficile: Role of secondary bile acids. Cell Chem. Biol. 2019, 26, 27–34. [Google Scholar] [CrossRef]
- Thanissery, R.; Winston, J.A.; Theriot, C.M. Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 2017, 45, 86–100. [Google Scholar] [CrossRef]
- Hegyi, P.; Maléth, J.; Walters, J.R.; Hofmann, A.F.; Keely, S.J. Guts and gall: Bile acids in regulation of intestinal epithelial function in health and disease. Physiol. Rev. 2018, 98, 1983–2023. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Gaitonde, S.V.; Qi, W.; Martinez, J.D. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis 2001, 22, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Tortora, K.; Vitali, F.; De Filippo, C.; Caderni, G.; Giovannelli, L. DNA damage in colon mucosa of Pirc rats, an Apc-driven model of colon tumorigenesis. Toxicol. Lett. 2020, 324, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Majumdar, A.P. Bile acid: A potential inducer of colon cancer stem cells. Stem. Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef]
- Kong, Y.; Bai, P.S.; Sun, H.; Nan, K.J.; Chen, N.Z.; Qi, X.G. The deoxycholic acid targets miRNA-dependent CAC1 gene expression in multidrug resistance of human colorectal cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2321–2332. [Google Scholar] [CrossRef]
- Guz, M.; Jeleniewicz, W.; Malm, A.; Korona-Glowniak, I. A Crosstalk between Diet, Microbiome and microRNA in Epigenetic Regulation of Colorectal Cancer. Nutrients 2021, 13, 2428. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).