
Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt

Abstract
:1. Introduction
2. Results
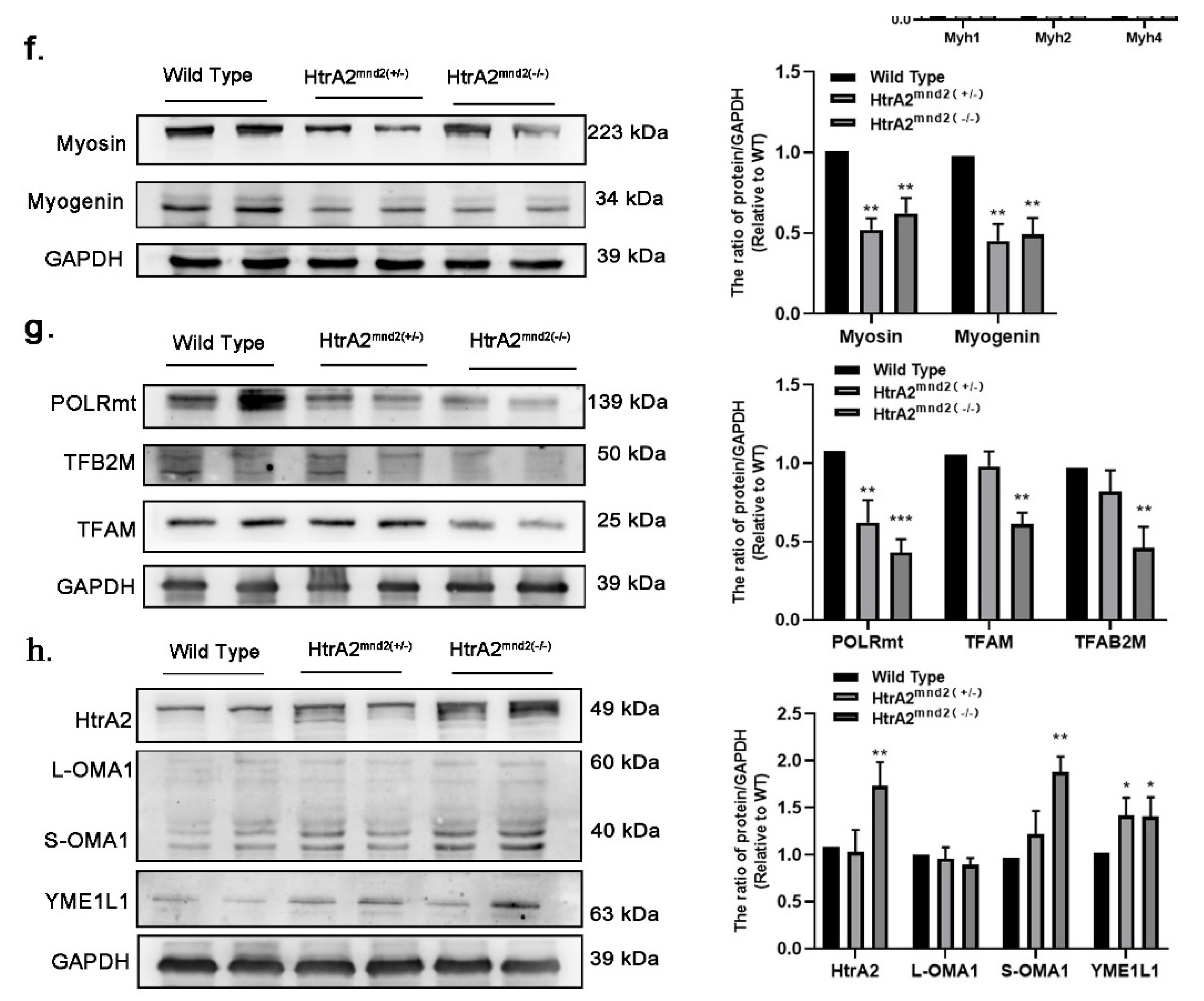
2.1. HtrA2/Omi Protease Deficiency Induces Sarcopenia in Mnd2 Mice
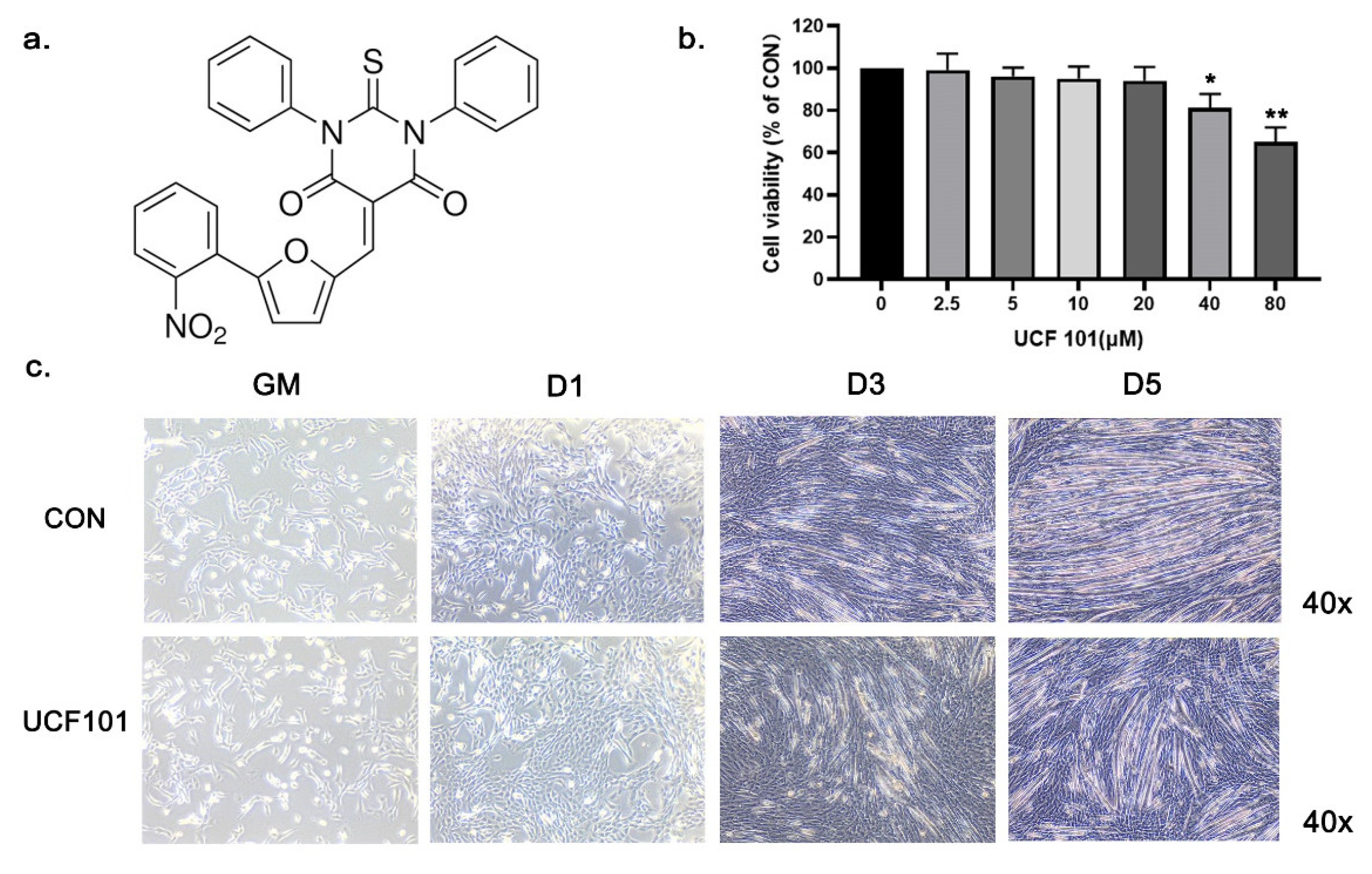
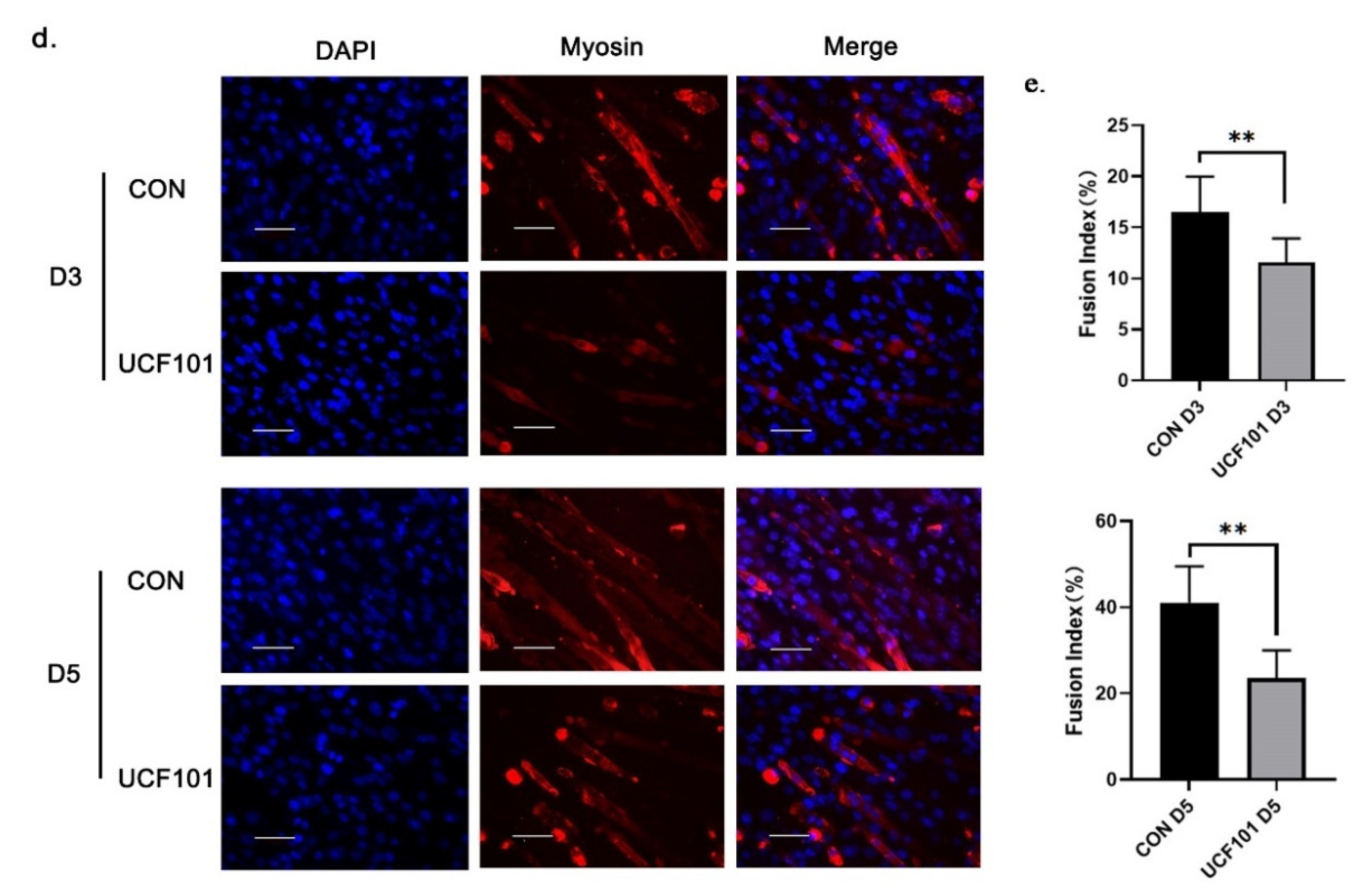
2.2. HtrA2/Omi Protease Deficiency Impairs the Differentiation of C2C12 Myoblast
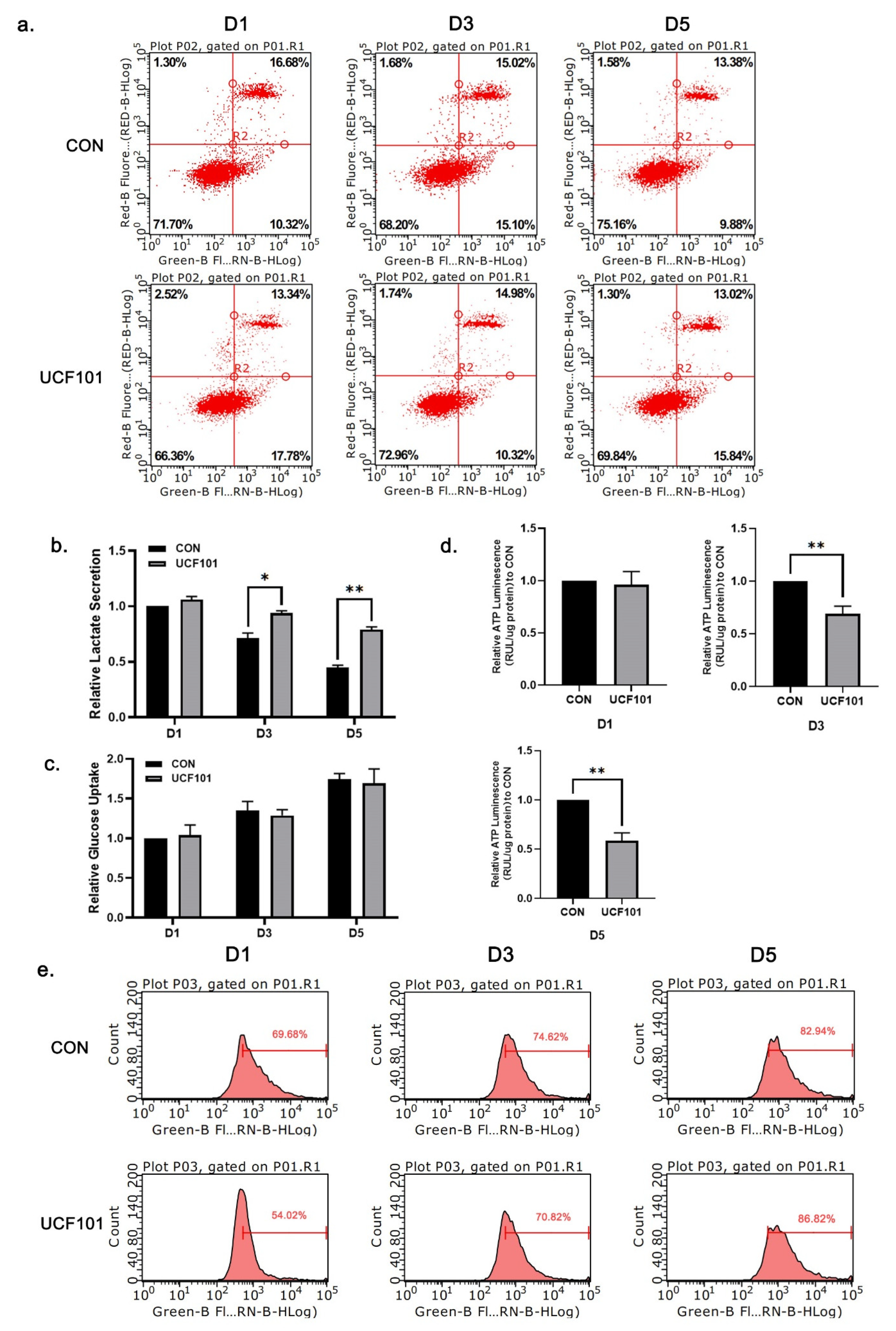
2.3. HtrA2/Omi Protease Deficiency Causes OXPHOS Hypofunction
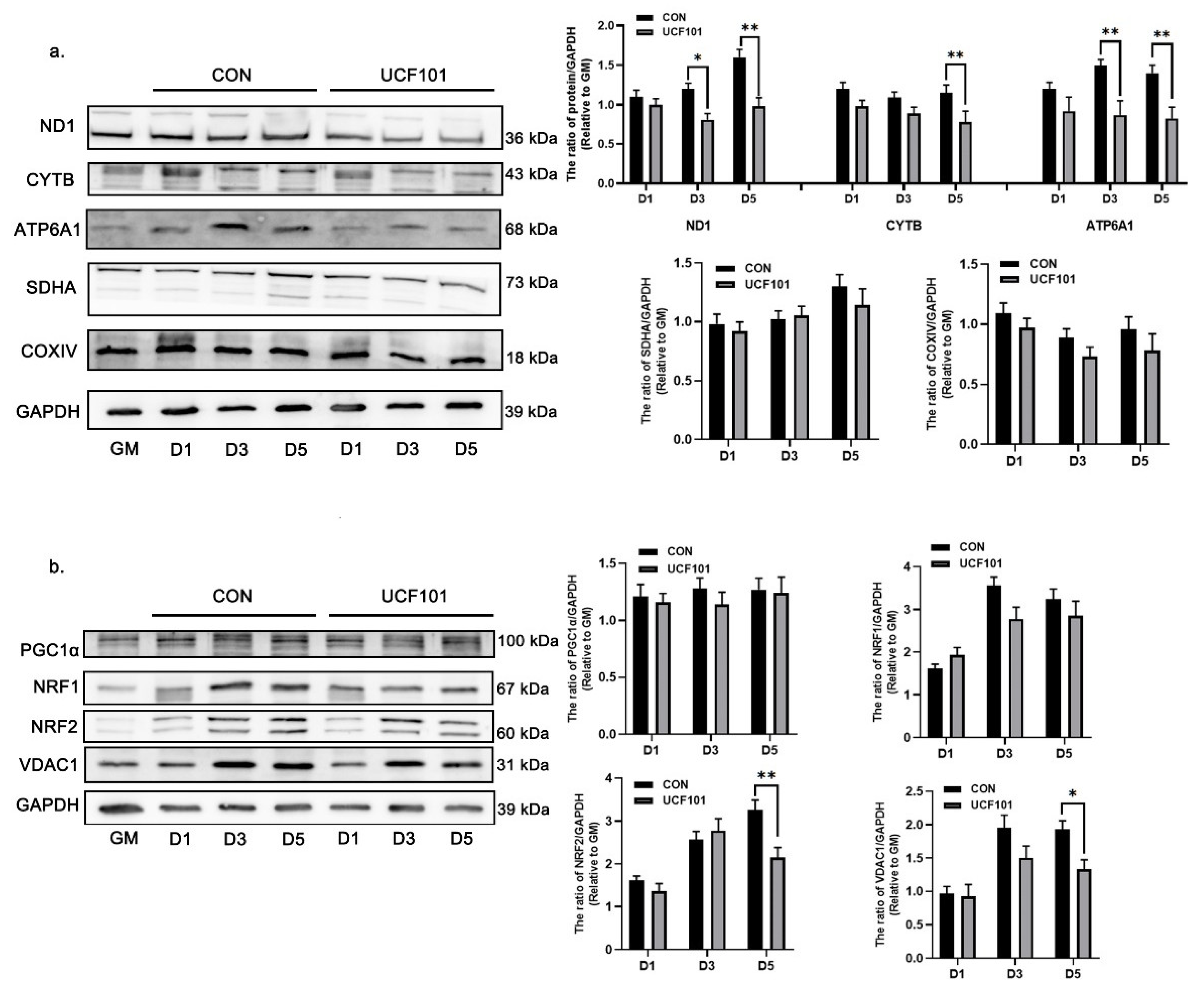
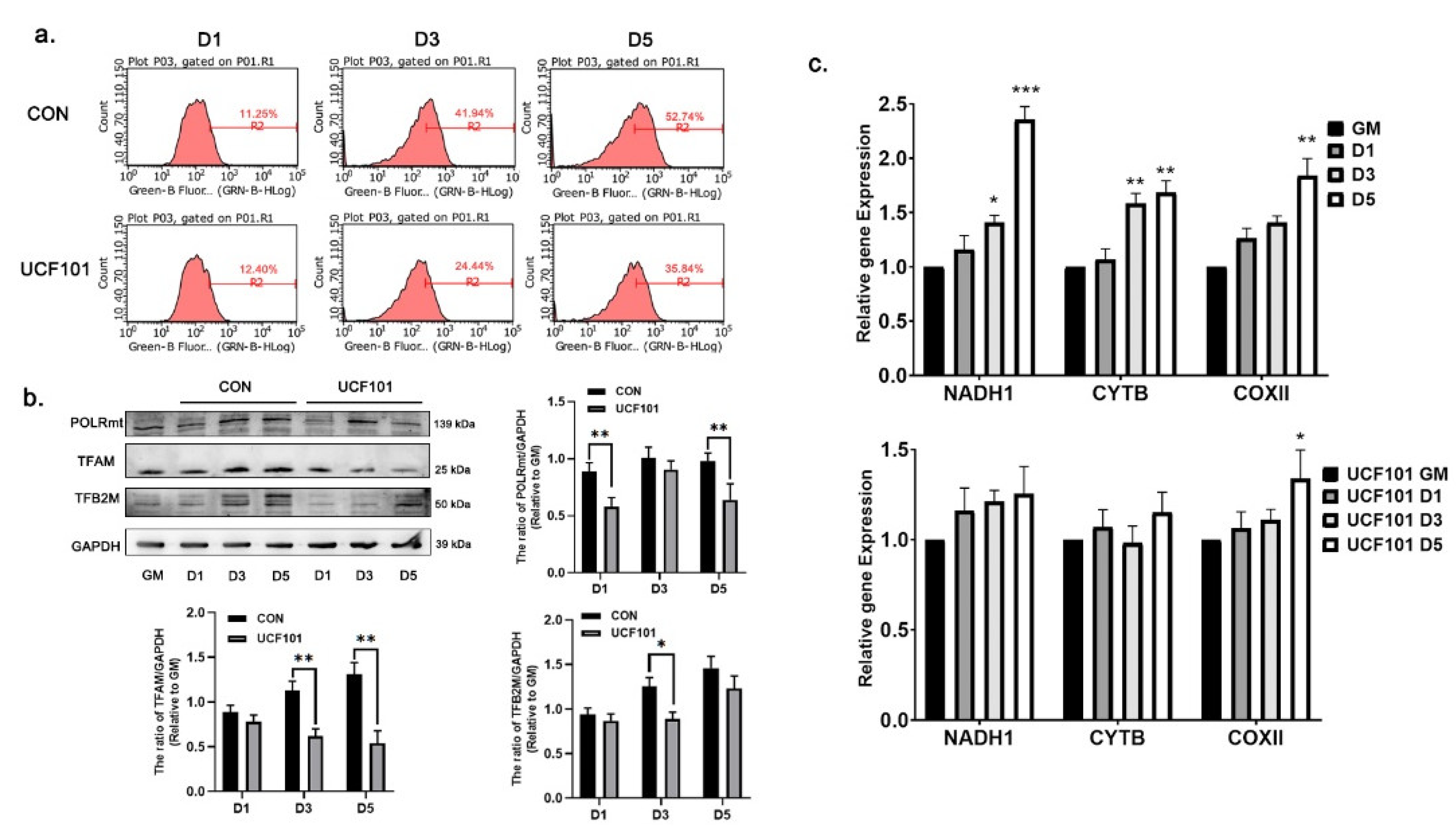
2.4. HtrA2/Omi Protease Deficiency Leads to Mitonuclear Imbalance
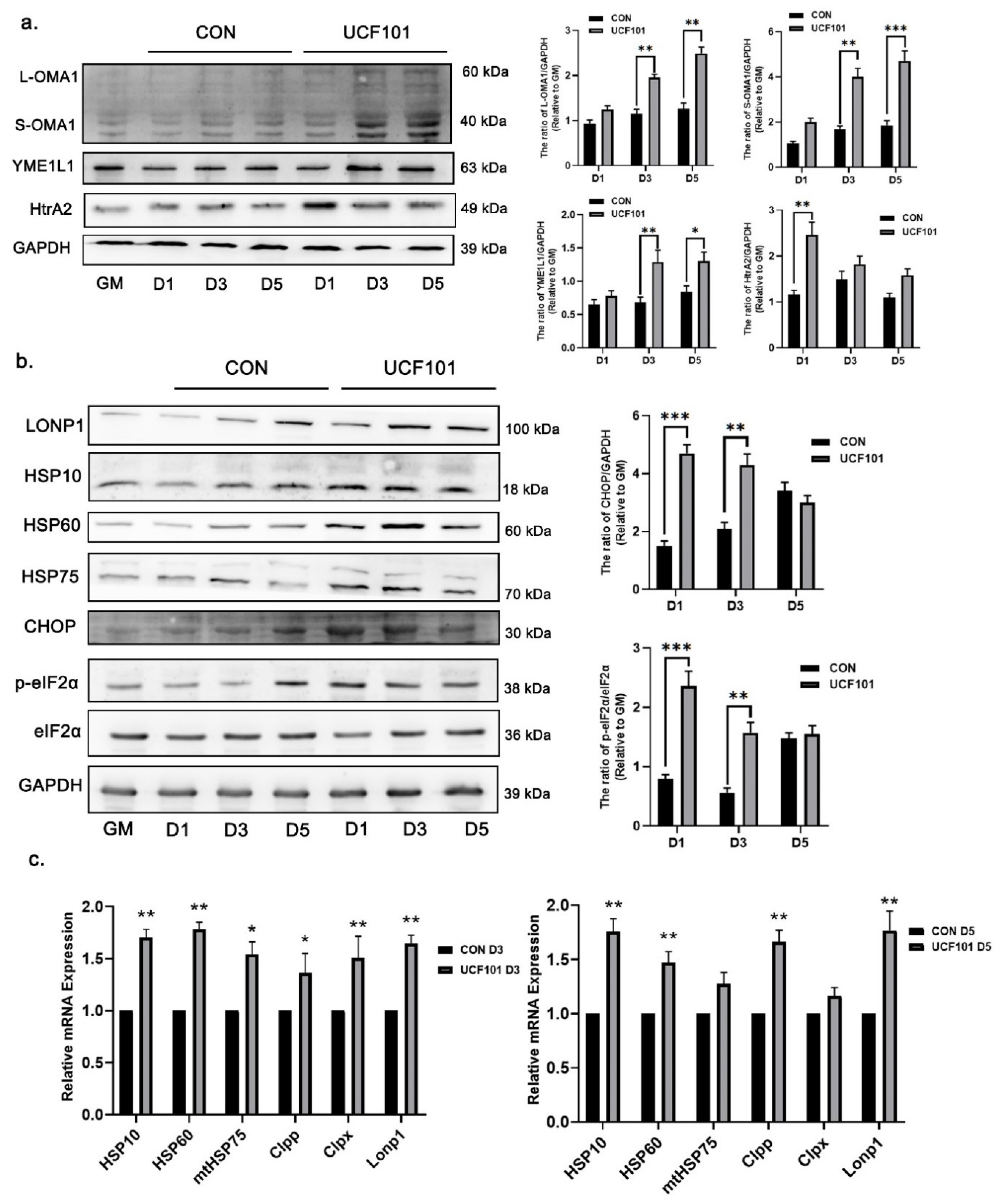
2.5. HtrA2/Omi Protease Deficiency Activates UPRmt during Myogenic Differentiation
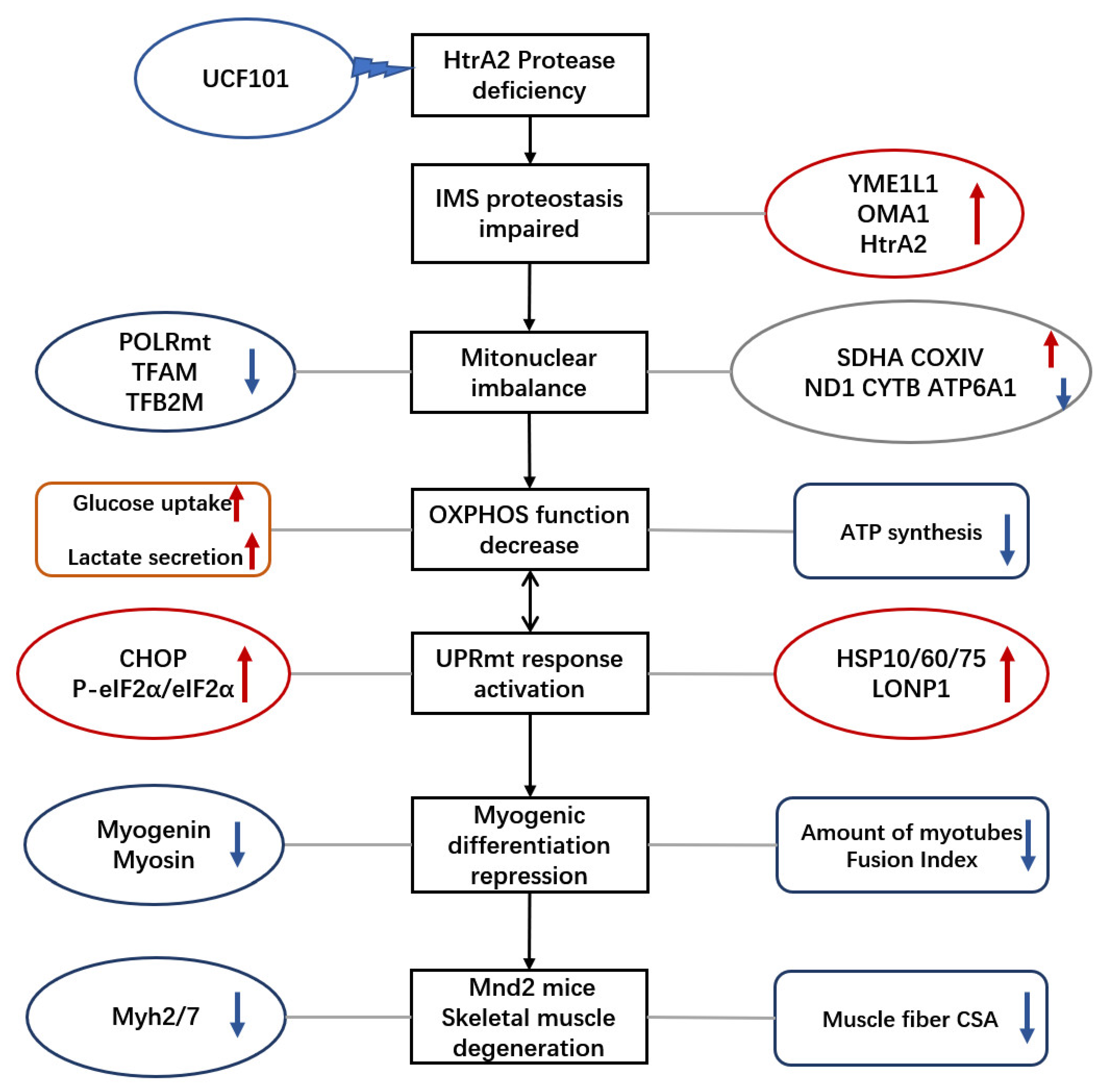
3. Discussion
4. Materials and Methods
4.1. Animals and Genotyping
4.2. Histological and Morphological Quantitative Analysis of Muscle Fibers
4.3. C2C12 Myoblast Culture and Differentiation
4.4. Measurement of Cell Viability
4.5. Immunofluorescence Microscopy
4.6. Quantitative Real-Time PCR
4.7. Western Blot Analysis
4.8. Quantification of Mitochondrial Copy Numbers
4.9. Analysis of Glucose Uptake and Lactate Secretion
4.10. Measurement of ATP Content
4.11. Flow Cytometry
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pratt, J.; Boreham, C.; Ennis, S.; Ryan, A.W.; De Vito, G. Genetic Associations with Aging Muscle: A Systematic Review. Cells 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, E.; Liu, Y.; Samuel, A.; Or, O.; Lane, J. A review of sarcopenia: Enhancing awareness of an increasingly prevalent disease. Bone 2017, 105, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Fielding, R.A.; Vellas, B.; Evans, W.J.; Bhasin, S.; Morley, J.E.; Newman, A.B.; van Kan, G.A.; Andrieu, S.; Bauer, J.; Breuille, D.; et al. Sarcopenia: An undiagnosed condition in older adults. Current consensus definition: Prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med. Dir. Assoc. 2011, 12, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochlin, K.; Yu, S.; Roy, S.; Baylies, M.K. Myoblast fusion: When it takes more to make one. Dev. Biol. 2010, 341, 66–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.E.; Suetta, C.; Conboy, M.J.; Aagaard, P.; Mackey, A.; Kjaer, M.; Conboy, I. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol. Med. 2009, 1, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 2013, 280, 3991–4003. [Google Scholar] [CrossRef]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Kim, J.-S.; Yoon, Y.; Santiago, M.C.; Brown, M.D.; Park, J.-Y. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R927–R938. [Google Scholar] [CrossRef]
- Rochard, P.; Rodier, A.; Casas, F.; Cassar-Malek, I.; Marchal-Victorion, S.; Daury, L.; Wrutniak, C.; Cabello, G. Mitochondrial activity is involved in the regulation of myoblast differentiation through myogenin expression and activity of myogenic factors. J. Biol. Chem. 2000, 275, 2733–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyer, P.; Grandemange, S.; Busson, M.; Carazo, A.; Gamaléri, F.; Pessemesse, L.; Casas, F.; Cabello, G.; Wrutniak-Cabello, C. Mitochondrial activity regulates myoblast differentiation by control of c-Myc expression. J. Cell. Physiol. 2006, 207, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, N.H.; Middelkoop, E.; Adorf, M.; Dekker, H.L.; Van Galen, M.J.; Berg, M.V.D.; Bolhuis, P.A.; Bogert, C.V.D. Mitochondria in cultured human muscle cells depleted of mitochondrial DNA. Eur. J. Cell. Biol. 1993, 61, 400–408. [Google Scholar] [PubMed]
- Brunk, C.; Yaffe, D. The reversible inhibition of myoblast fusion by ethidium bromide (EB). Exp. Cell Res. 1976, 99, 310–318. [Google Scholar] [CrossRef]
- Wagatsuma, A.; Kotake, N.; Yamada, S. Muscle regeneration occurs to coincide with mitochondrial biogenesis. Mol. Cell. Biochem. 2011, 349, 139–147. [Google Scholar] [CrossRef]
- Korohoda, W.; Pietrzkowski, Z.; Reiss, K. Chloramphenicol, an inhibitor of mitochondrial protein synthesis, inhibits myoblast fusion and myotube differentiation. Folia Histochem. Cytobiol. 1993, 31, 9–13. [Google Scholar]
- Zhou, H.; Yuan, D.; Gao, W.; Tian, J.; Sun, H.; Yu, S.; Wang, J.; Sun, L. Loss of high-temperature requirement protein A2 protease activity induces mitonuclear imbalance via differential regulation of mitochondrial biogenesis in sarcopenia. IUBMB Life 2020, 72, 1659–1679. [Google Scholar] [CrossRef]
- Leary, S.C.; Battersby, B.J.; Hansford, R.G.; Moyes, C.D. Interactions between bioenergetics and mitochondrial biogenesis. Biochim. Biophys. Acta 1998, 1365, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Ljubicic, V.; Hood, D.A. Diminished contraction-induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell 2009, 8, 394–404. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Richter-Dennerlein, R.; Pacheu-Grau, D.; Liu, F.; Zhu, Y.; Dennerlein, S.; Rehling, P. MITRAC15/COA1 promotes mitochondrial translation in a ND2 ribosome-nascent chain complex. EMBO Rep. 2020, 21, e48833. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsör, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483.e10. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.; Saunders, J.M.; Wiseman, R.L. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; Van Der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.W.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorko-Glonek, J.; Zurawa-Janicka, D.; Koper, T.; Jarzab, M.; Figaj, D.; Glaza, P.; Lipinska, B. HtrA protease family as therapeutic targets. Curr. Pharm. Des. 2013, 19, 977–1009. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Datta, P.; Srinivasula, S.M.; Ji, W.; Gupta, S.; Zhang, Z.; Davies, E.; Hajnóczky, G.; Saunders, T.; Van Keuren, M.L.; et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003, 425, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.; Yu, H.; Kong, Q.; Wang, B.; Shen, L.; Dong, D.; Sun, L. The Mitochondrial PHB2/OMA1/DELE1 Pathway Cooperates with Endoplasmic Reticulum Stress to Facilitate the Response to Chemotherapeutics in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 1320. [Google Scholar] [CrossRef] [PubMed]
- Cilenti, L.; Lee, Y.; Hess, S.; Srinivasula, S.; Park, K.M.; Junqueira, D.; Davis, H.; Bonventre, J.V.; Alnemri, E.S.; Zervos, A.S. Characterization of a novel and specific inhibitor for the pro-apoptotic protease Omi/HtrA2. J. Biol. Chem. 2003, 278, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Schmelter, C.; Fomo, K.N.; Perumal, N.; Pfeiffer, N.; Grus, F.H. Regulation of the HTRA2 Protease Activity by an Inhibitory Antibody-Derived Peptide Ligand and the Influence on HTRA2-Specific Protein Interaction Networks in Retinal Tissues. Biomedicines 2021, 9, 1013. [Google Scholar] [CrossRef]
- Klupsch, K.; Downward, J. The protease inhibitor Ucf-101 induces cellular responses independently of its known target, HtrA2/Omi. Cell Death Differ. 2006, 13, 2157–2159. [Google Scholar] [CrossRef]
- Schiaffino, S. Muscle fiber type diversity revealed by anti-myosin heavy chain antibodies. FEBS J. 2018, 285, 3688–3694. [Google Scholar] [CrossRef]
- Venuti, J.M.; Morris, J.H.; Vivian, J.L.; Olson, E.N.; Klein, W.H. Myogenin is required for late but not early aspects of myogenesis during mouse development. J. Cell Biol. 1995, 128, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, A.; Liu, S.; He, Q.; Luo, Y.; He, Z.; Chen, Y.; Tao, A.; Yan, J. Inhibition of HtrA2 alleviated dextran sulfate sodium (DSS)-induced colitis by preventing necroptosis of intestinal epithelial cells. Cell Death Dis. 2019, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell. 2019, 73, 1028–1043.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Beckervordersandforth, R.; Ebert, B.; Schäffner, I.; Moss, J.; Fiebig, C.; Shin, J.; Moore, D.L.; Ghosh, L.; Trinchero, M.F.; Stockburger, C.; et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron 2017, 93, 560–573.e6. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.-E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Remels, A.; Langen, R.; Schrauwen, P.; Schaart, G.; Schols, A.; Gosker, H. Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell. Endocrinol. 2010, 315, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Prosser, R.; Simonetti, S.; Sadlock, J.; Jagiello, G.; Schon, E.A. Rearranged mitochondrial genomes are present in human oocytes. Am. J. Hum. Genet. 1995, 57, 239–247. [Google Scholar]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [Green Version]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular mechanisms of apoptosis in cerebral ischemia: Multiple neuroprotective opportunities. Mol. Neurobiol. 2008, 37, 7–38. [Google Scholar] [CrossRef] [PubMed]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Goo, H.G.; Rhim, H.; Kang, S. Pathogenic Role of Serine Protease HtrA2/Omi in Neurodegenerative Diseases. Curr. Protein Pept. Sci. 2017, 18, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wang, X.; Yu, J.; Tian, Y.; Yang, C.; Chen, Y.; Chen, H.; Ge, H. LonP1 regulates mitochondrial network remodeling through the PINK1/Parkin pathway during myoblast differentiation. Am. J. Physiol. Cell Physiol. 2020, 319, C1020–C1028. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic. Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Alter, J.; Bengal, E. Stress-induced C/EBP homology protein (Chop) represses MyoD transcription to delay myoblast differentiation. PLoS ONE 2011, 6, e29498. [Google Scholar] [CrossRef] [Green Version]
- Bolus, D.J.; Shanmugam, G.; Narasimhan, M.; Rajasekaran, N.S. Recurrent heat shock impairs the proliferation and differentiation of C2C12 myoblasts. Cell Stress Chaperones 2018, 23, 399–410. [Google Scholar] [CrossRef]
- Desgeorges, T.; Liot, S.; Lyon, S.; Bouvière, J.; Kemmel, A.; Trignol, A.; Rousseau, D.; Chapuis, B.; Gondin, J.; Mounier, R.; et al. Open-CSAM, a new tool for semi-automated analysis of myofiber cross-sectional area in regenerating adult skeletal muscle. Skelet. Muscle 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Hur, W.; Kang, B.Y.; Kim, S.M.; Lee, G.W.; Kim, J.H.; Nam, M.K.; Rhim, H.; Yoon, S.K. Serine Protease HtrA2/Omi Deficiency Impairs Mitochondrial Homeostasis and Promotes Hepatic Fibrogenesis via Activation of Hepatic Stellate Cells. Cells 2019, 8, 1119. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers 5′-3′ | ||
|---|---|---|
| Gene | Forward | Reverse |
| Mnd2 | CACACTGAGGATTCAAACCAAGGT | GACCGAGGACATAAACAGGGTGTA |
| Primers 5′-3′ | ||
|---|---|---|
| Gene | Forward | Reverse |
| GAPDH | GTCGTGGAGTCTACTGGTGTC | GAGCCCTTCCACAATGCCAAA |
| MyoD | GACAGGGAGGAGGGGTAGAG | TGCTGTCTCAAAGGAGCAGA |
| myogenin | CCTACAGACGCCCACAATCT | CAGGGCTGTTTTCTGGACAT |
| Myh1 | CTCTTCCCGCTTTGGTAAGTT | CAGGAGCATTTCGATTAGATCCG |
| Myh2 | GCACCCATCCTCATTTCGTGA | GGAATGGCACTTGCGTTTAACA |
| Myh4 | CTTTGCTTACGTCAGTCAAGGT | AGCGCCTGTGAGCTTGTAAA |
| Myh7 | ACTGTCAACACTAAGAGGGTCA | TTGGATGATTTGATCTTCCAGGG |
| Lonp1 | CTCATGGTGGAGGTTGAGAATG | CAGAGGGTTCAAGGCGATGAT |
| Clpp | CCATCTACGACACAATGCAGT | GAATTGGGCAGTGAATGGCG |
| Clpx | CCAGGCTGGATATGTAGGTGA | GCACACTGCCAATCTTATCTACT |
| Chop | AAGCCTGGTATGAGGATCTGC | TTCCTGGGGATGAGATATAGGTG |
| Hsp10 | TTCCGCTCTTTGACAGAGTATTG | TCTGGAAGCATAATGCCACCT |
| Hsp60 | GCCTTAATGCTTCAAGGTGTAGA | CCCCATCTTTTGTTACTTTGGGA |
| HSP75 | CAGGACAGTTATACAGCACACAG | TACAGAGAACGGGCTACGATG |
| Primers 5′-3′ | ||
|---|---|---|
| Gene | Forward | Reverse |
| β-globin | GAAGCGATTCTAGGGAGCAG | GGAGCAGCGATTCTGAGTAGA |
| mNADH1 | TCTGCCAGCCTGACCCATAG | CCGGCTGCGTATTCTACGTT |
| mCOXII | GCCGACTAAATCAAGCAACA | CAATGGGCATAAAGCTATGG |
| mCYTB | TATTCCTTCATGTCGGACGA | AAATGCTGTGGCTATGACTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Shen, L.; Zhang, P.; Lin, F.; Ma, J.; Wu, Y.; Yu, H.; Sun, L. Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt. Int. J. Mol. Sci. 2022, 23, 11761. https://doi.org/10.3390/ijms231911761
Sun H, Shen L, Zhang P, Lin F, Ma J, Wu Y, Yu H, Sun L. Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt. International Journal of Molecular Sciences. 2022; 23(19):11761. https://doi.org/10.3390/ijms231911761
Chicago/Turabian StyleSun, Hongyu, Luyan Shen, Ping Zhang, Fu Lin, Jiaoyan Ma, Ying Wu, Huimei Yu, and Liankun Sun. 2022. "Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt" International Journal of Molecular Sciences 23, no. 19: 11761. https://doi.org/10.3390/ijms231911761
APA StyleSun, H., Shen, L., Zhang, P., Lin, F., Ma, J., Wu, Y., Yu, H., & Sun, L. (2022). Inhibition of High-Temperature Requirement Protein A2 Protease Activity Represses Myogenic Differentiation via UPRmt. International Journal of Molecular Sciences, 23(19), 11761. https://doi.org/10.3390/ijms231911761