
The Epitranscriptomic Mechanism of Metal Toxicity and Carcinogenesis

{kind=link}
Abstract
:1. Introduction
2. Epitranscriptomics
2.1. A Brief History of Epitranscriptomics
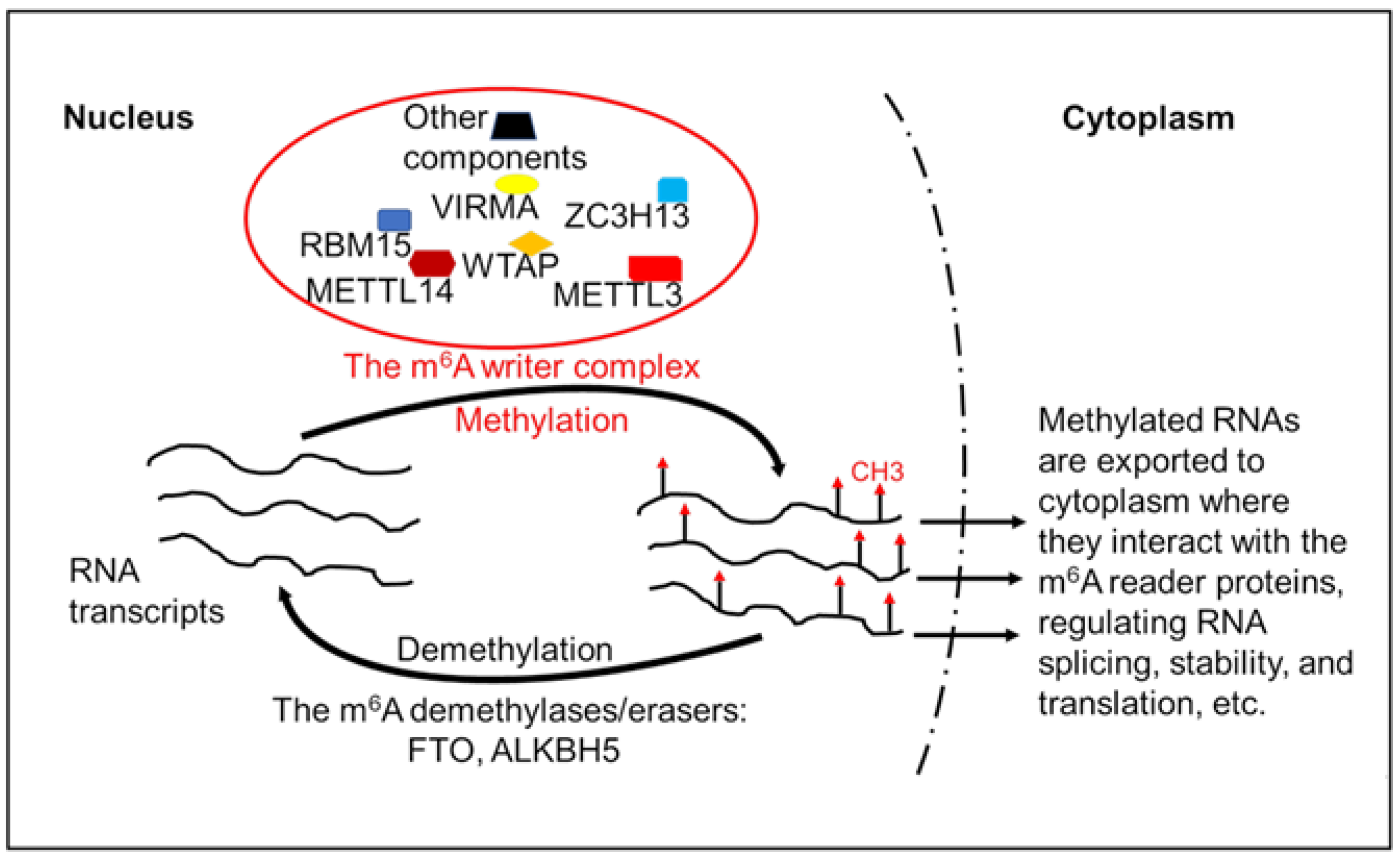
2.2. Modifiers and Regulators of the Epitranscriptome
2.3. Functions of Epitranscriptomic Regulations
3. Epitranscriptomic Mechanisms of Metal Toxicity and Carcinogenesis
3.1. Arsenic
3.1.1. Epitranscriptomic Mechanism of Arsenic Toxicity
3.1.2. Epitranscriptomic Mechanism of Arsenic Carcinogenesis
3.2. Cadmium
3.2.1. Epitranscriptomic Mechanism of Cadmium Toxicity
3.2.2. Epitranscriptomic Mechanism of Cadmium Carcinogenesis
3.3. Chromium
3.3.1. Epitranscriptomic Mechanism of Chromium Toxicity
3.3.2. Epitranscriptomic Mechanism of Chromium Carcinogenesis
4. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| A3B | APOBEC3B |
| Akt | protein kinase B |
| ALKBH5 | alkB homologue 5 |
| AS3MT | arsenite methyltransferase |
| ATSDR | Agency for Toxic Substances and Disease Registry |
| CAT | catalase |
| Cr | chromium |
| FTO | fat mass and obesity-associated protein |
| HaCaT | human keratinocyte |
| Jak | Janus kinase |
| m6A | N6-methyladenosine |
| MAPK | Mitogen-activated protein kinase |
| METTL14 | methyltransferase like-14 |
| METTL3 | methyltransferase like-3 |
| MNT | MAX network transcriptional repressor |
| mRNA | messenger ribonucleic acid |
| mTOT | mammalian target of rapamycin |
| NAC | N-acetylcysteine |
| NEDD4L | NEDD4-like E3 ubiquitin protein ligase |
| NLRP3 | NOD-like receptor protein 3 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| PI3K | phosphatidylinositol-3-kinase |
| PTM | post-translational modification |
| ROS | reactive oxygen species |
| rRNA | ribosomal RNA |
| SOD | superoxide dismutase |
| STAT | signal transducer and activator of transcription |
| tRNA | transfer ribonucleic acid |
| UVB | ultraviolet B light (wavelength between 280–315 nm) |
| WTAP | Wilms’ tumor 1-associating protein |
| YTHDC1-2 | YTH domain-containing proteins1–2 |
| YTHDF1-3 | YTH domain-containing family proteins1–3 |
References
- Sun, Q.; Li, Y.; Shi, L.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 2022, 469, 153136. [Google Scholar] [CrossRef] [PubMed]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury; Lead; Chromium; Cadmium; and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.S.; Wang, Z.; Yang, C. Dysregulations of long non-coding RNAs—The emerging “lnc” in environmental carcinogenesis. Semin. Cancer Biol. 2021, 76, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kocadal, K.; Alkas, F.B.; Battal, D.; Saygi, S. Cellular pathologies and genotoxic effects arising secondary to heavy metal exposure: A review. Hum. Exp. Toxicol. 2020, 39, 3–13. [Google Scholar] [CrossRef]
- Zhu, Y.; Costa, M. Metals and molecular carcinogenesis. Carcinogenesis 2020, 41, 1161–1172. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, C. Metal carcinogen exposure induces cancer stem cell-like property through epigenetic reprograming: A novel mechanism of metal carcinogenesis. Semin. Cancer Biol. 2019, 57, 95–104. [Google Scholar] [CrossRef]
- Humphries, B.; Wang, Z.; Yang, C. The role of microRNAs in metal carcinogen-induced cell malignant transformation and tumorigenesis. Food Chem. Toxicol. 2016, 98 Pt A, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Moshitch-Moshkovitz, S.; Dominissini, D.; Rechavi, G. The epitranscriptome toolbox. Cell 2022, 185, 764–776. [Google Scholar] [CrossRef]
- Primac, I.; Penning, A.; Fuks, F. Cancer epitranscriptomics in a nutshell. Curr. Opin. Genet. Dev. 2022, 75, 101924. [Google Scholar] [CrossRef]
- Yang, C. ToxPoint: Dissecting Functional RNA Modifications in Responses to Environmental Exposure-Mechanistic Toxicology Research Enters a New Era. Toxicol. Sci. 2020, 174, 1–2. [Google Scholar] [CrossRef]
- Roundtree, I.A.; He, C. RNA epigenetics–chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 2016, 30, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Bagiński, B. MODOMICS: An Operational Guide to the Use of the RNA Modification Pathways Database. Methods Mol. Biol. 2021, 2284, 481–505. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Bokar, J.A.; Rath-Shambaugh, M.E.; Ludwiczak, R.; Narayan, P.; Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994, 269, 17697–17704. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Jia, G.F.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.Q.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wei, J.; He, C. Where; When; and How: Context-Dependent Functions of RNA Methylation Writers; Readers; and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic Transcriptomic m6A Decoration: Writers; Erasers; Readers and Functions in RNA Metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading; Writing and Erasing mRNA Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.B.; Wang, Z.; Yang, C. The m6A RNA methylation regulates oncogenic signaling pathways driving cell malignant transformation and carcinogenesis. Mol. Cancer 2021, 20, 61. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.B.; Wang, Z.; Yang, C. Roles of m6A RNA Modification in Normal Development and Disease. RNA Technol. 2021, 12, 267–308. [Google Scholar]
- Uddin, M.B.; Wang, Z.; Yang, C. Dysregulations of Functional RNA Modifications in Cancer; Cancer Stemness and Cancer Therapeutics. Theranostics 2020, 10, 3164–3189. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Das, A.S.; Alfonzo, J.D.; Accornero, F. The importance of RNA modifications: From cells to muscle physiology. Wiley Interdiscip. Rev. RNA 2022, 13, e1700. [Google Scholar] [CrossRef]
- Dong, L.; Cao, Y.; Hou, Y.; Liu, G. N6 -methyladenosine RNA methylation: A novel regulator of the development and function of immune cells. J. Cell Physiol. 2022, 237, 329–345. [Google Scholar] [CrossRef]
- Ilieva, M.; Uchida, S. Functional roles of epitranscriptomic marks in the cardiovascular system and disease: A narrative review. Ann. Transl. Med. 2022, 10, 753. [Google Scholar] [CrossRef]
- Sokpor, G.; Xie, Y.; Nguyen, H.P.; Tuoc, T. Emerging Role of m6 A Methylome in Brain Development: Implications for Neurological Disorders and Potential Treatment. Front. Cell Dev. Biol. 2021, 9, 656849. [Google Scholar] [CrossRef] [PubMed]
- Berulava, T.; Buchholz, E.; Elerdashvili, V.; Pena, T.; Islam, M.R.; Lbik, D.; Mohamed, B.A.; Renner, A.; von Lewinski, D.; Sacherer, M.; et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Jiapaer, Z.; Su, D.; Hua, L.; Lehmann, H.I.; Gokulnath, P.; Vulugundam, G.; Song, S.; Zhang, L.; Gong, Y.; Li, G. Regulation and roles of RNA modifications in aging-related diseases. Aging Cell 2022, 21, e13657. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Siegal, E.; Gregory, R.I. tRNA dysregulation and disease. Nat. Rev. Genet. 2022; 9, online ahead of print. [Google Scholar] [CrossRef]
- Kurkowiak, M.; Arcimowicz, Ł.; Chruściel, E.; Urban-Wójciuk, Z.; Papak, I.; Keegan, L.; O’Connell, M.; Kowalski, J.; Hupp, T.; Marek-Trzonkowska, N. The effects of RNA editing in cancer tissue at different stages in carcinogenesis. RNA Biol. 2021, 18, 1524–1539. [Google Scholar] [CrossRef]
- Xue, C.; Chu, Q.; Zheng, Q.; Jiang, S.; Bao, Z.; Su, Y.; Lu, J.; Li, L. Role of main RNA modifications in cancer: N 6-methyladenosine; 5-methylcytosine; and pseudouridine. Signal Transduct. Target Ther. 2022, 7, 142. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, Y.; Wu, P.; Zi, X.; Sun, N.; He, J. The potential role of N 7-methylguanosine (m7G) in cancer. J. Hematol. Oncol. 2022, 15, 63. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, Y.; Jin, L.; Yang, F.; Ding, H.; Zhang, L.; Li, L.; Pan, T. The Emerging Role of N6-Methyladenosine RNA Methylation as Regulators in Cancer Therapy and Drug Resistance. Front. Pharmacol. 2022, 13, 873030. [Google Scholar] [CrossRef]
- Cerneckis, J.; Cui, Q.; He, C.; Yi, C.; Shi, Y. Decoding pseudouridine: An emerging target for therapeutic development. Trends Pharmacol. Sci. 2022, 43, 522–535. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Deng, Y.; Yi, P.; Yu, J. Targeting the RNA m 6 A modification for cancer immunotherapy. Mol. Cancer 2022, 21, 76. [Google Scholar] [CrossRef]
- Dang, Q.; Shao, B.; Zhou, Q.; Chen, C.; Guo, Y.; Wang, G.; Liu, J.; Kan, Q.; Yuan, W.; Sun, Z. RNA N 6-Methyladenosine in Cancer Metastasis: Roles; Mechanisms; and Applications. Front. Oncol. 2021, 11, 681781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, F.; Chen, W.; Miao, H.; Liang, H.; Liao, Z.; Zhang, Z.; Zhang, B. The role of RNA m5C modification in cancer metastasis. Int. J. Biol. Sci. 2021, 17, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Heras, C.R.; Lee, G. m6A in the Signal Transduction Network. Mol. Cells 2022, 45, 435–443. [Google Scholar] [CrossRef]
- Liu, F.; Su, X. Effects of m6A modifications on signaling pathways in human cancer (Review). Oncol. Rep. 2021, 45, 36. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Ye, M.; Liu, B.; Wei, M.; Ma, D.; Dong, K. m6A Modification: A Double-Edged Sword in Tumor Development. Front. Oncol. 2021, 11, 679367. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.; Cui, Y.H.; He, Y.Y. Context-Dependent Roles of RNA Modifications in Stress Responses and Diseases. Int. J. Mol. Sci. 2021, 22, 1949. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, J.; Wang, X.; Ying, Y.; Xie, H.; Yan, H.; Zheng, X.; Xie, L. The dual role of N6-methyladenosine modification of RNAs is involved in human cancers. J. Cell Mol. Med. 2018, 22, 4630–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ATSDR (Agency for Toxic Substances and Disease Research). Top 20 Hazardous Substances: ATSDR/EPA Priority List for 2017; U.S. Department of Health and Human Services Public Health Service/U.S. Environmental Protection Agency: Washington, DC, USA, 2019. Available online: https://www.atsdr.cdc.gov/SPL/index.html#2019spl (accessed on 8 August 2022).
- Martínez-Castillo, M.; García-Montalvo, E.A.; Arellano-Mendoza, M.G.; Sánchez-Peña, L.D.C.; Soria Jasso, L.E.; Izquierdo-Vega, J.A.; Valenzuela, O.L.; Hernández-Zavala, A. Arsenic exposure and non-carcinogenic health effects. Hum. Exp. Toxicol. 2021, 40 (Suppl. 12), S826–S850. [Google Scholar] [CrossRef]
- Escudero-Lourdes, C. Toxicity mechanisms of arsenic that are shared with neurodegenerative diseases and cognitive impairment: Role of oxidative stress and inflammatory responses. Neurotoxicology 2016, 53, 223–235. [Google Scholar] [CrossRef]
- Danes, J.M.; Palma, F.R.; Bonini, M.G. Arsenic and other metals as phenotype driving electrophiles in carcinogenesis. Semin. Cancer Biol. 2021, 76, 287–291. [Google Scholar] [CrossRef]
- Signes-Pastor, A.J.; Romano, M.E.; Jackson, B.; Braun, J.M.; Yolton, K.; Chen, A.; Lanphear, B.; Karagas, M.R. Associations of maternal urinary arsenic concentrations during pregnancy with childhood cognitive abilities: The HOME study. Int. J. Hyg. Environ. Health 2022, 245, 114009. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Tang, Q.; Zou, Z.; Meng, P.; Tu, B.; Xia, Y.; Cheng, S.; Zhang, L.; Yang, K.; Mu, S.; et al. m6A Demethylase FTO Regulates Dopaminergic Neurotransmission Deficits Caused by Arsenite. Toxicol. Sci. 2018, 165, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.E.; Hess, S.; Meyer, K.D.; Verhagen, L.A.W.; Koch, L.; Bronneke, H.S.; Dietrich, M.O.; Jordan, S.D.; Saletore, Y.; Elemento, O.; et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci. 2013, 16, 1042. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, T.; Sun, D.; Wu, M.; Zhang, Z. Changes of RNA N6-methyladenosine in the hormesis effect induced by arsenite on human keratinocyte cells. Toxicol. Vitr. 2019, 56, 84–92. [Google Scholar] [CrossRef]
- Zhao, T.; Li, X.; Sun, D.; Zhang, Z. Oxidative stress: One potential factor for arsenite-induced increase of N6-methyladenosine in human keratinocytes. Environ. Toxicol. Pharmacol. 2019, 69, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wu, C.; Yao, X.; Han, Q.; Wang, N.; Yuan, W.; Zhang, J.; Shi, Y.; Jiang, L.; Liu, X.; et al. AS3MT facilitates NLRP3 inflammasome activation by m6A modification during arsenic-induced hepatic insulin resistance. Cell Biol. Toxicol. 2022, 28, 1–17. [Google Scholar] [CrossRef]
- Gu, S.; Sun, D.; Dai, H.; Zhang, Z. N6-methyladenosine mediates the cellular proliferation and apoptosis via microRNAs in arsenite-transformed cells. Toxicol. Lett. 2018, 292, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Sun, D.; Zhao, M.; Lai, Y.; Liu, Y.; Zhang, Z. N6-methyladenosine mediates arsenite-induced human keratinocyte transformation by suppressing p53 activation. Environ. Pollut. 2020, 259, 113908. [Google Scholar] [CrossRef]
- Cui, Y.H.; Yang, S.; Wei, J.; Shea, C.R.; Zhong, W.; Wang, F.; Shah, P.; Kibriya, M.G.; Cui, X.; Ahsan, H.; et al. Autophagy of the m6A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat. Commun. 2021, 12, 2183. [Google Scholar] [CrossRef]
- Gao, M.; Qi, Z.; Feng, W.; Huang, H.; Xu, Z.; Dong, Z.; Xu, M.; Han, J.; Kloeber, J.A.; Huang, J.; et al. m6A demethylation of cytidine deaminase APOBEC3B mRNA orchestrates arsenic-induced mutagenesis. J. Biol. Chem. 2022, 298, 101563. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef] [PubMed]
- Moghadamnia, A.A. Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135–145. [Google Scholar] [CrossRef]
- Ding, H.; Li, Z.; Li, X.; Yang, X.; Zhao, J.; Guo, J.; Lu, W.; Liu, H.; Wang, J. FTO Alleviates CdCl2-Induced Apoptosis and Oxidative Stress via the AKT/Nrf2 Pathway in Bovine Granulosa Cells. Int. J. Mol. Sci. 2022, 23, 4948. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, G.; Li, M.; Wang, L.; He, Z.; Gu, S. Study on the Correlation Between Regulatory Proteins of N6-methyladenosine and Oxidative Damage in Cadmium-induced Renal Injury. Biol. Trace Elem. Res. 2022, 6. online ahead of print. [Google Scholar] [CrossRef]
- Qu, T.; Mou, Y.; Dai, J.; Zhang, X.; Li, M.; Gu, S.; He, Z. Changes and relationship of N6-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicol. Lett. 2021, 343, 56–66. [Google Scholar] [CrossRef]
- Wu, B.; Jiang, X.; Huang, Y.; Ying, X.; Zhang, H.; Liu, B.; Li, Z.; Qi, D.; Ji, W.; Cai, X. Integrated analysis of mRNA-m 6 A-protein profiles reveals novel insights into the mechanisms for cadmium-induced urothelial transformation. Biomarkers 2021, 26, 499–507. [Google Scholar] [CrossRef]
- Li, L.; Zhou, M.; Chen, B.; Wang, Q.; Pan, S.; Hou, Y.; Xia, J.; Zhou, X. ALKBH5 promotes cadmium-induced transformation of human bronchial epithelial cells by regulating PTEN expression in an m6A-dependent manner. Ecotoxicol. Environ. Saf. 2021, 224, 112686. [Google Scholar] [CrossRef]
- Shi, H.; Hudson, L.G.; Liu, K.J. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic. Biol. Med. 2004, 37, 582–593. [Google Scholar] [CrossRef]
- Zhitkovich, A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem. Res. Toxicol. 2005, 18, 3–11. [Google Scholar] [CrossRef]
- Wise, S.S.; Holmes, A.L.; Wise, J.P., Sr. Hexavalent chromium-induced DNA damage and repair mechanisms. Rev. Environ. Health 2008, 23, 39–57. [Google Scholar] [CrossRef]
- Yao, H.; Guo, L.; Jiang, B.H.; Luo, J.; Shi, X. Oxidative stress and chromium(VI) carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 2008, 27, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Nickens, K.P.; Patierno, S.R.; Ceryak, S. Chromium genotoxicity: A double-edged sword. Chem. Biol. Interact. 2010, 188, 276–288. [Google Scholar] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [PubMed]
- Chervona, Y.; Arita, A.; Costa, M. Carcinogenic metals and the epigenome: Understanding the effect of nickel; arsenic; and chromium. Metallomics 2012, 4, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Brocato, J.; Costa, M. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit. Rev. Toxicol. 2013, 43, 493–514. [Google Scholar]
- Chen, Q.Y.; Murphy, A.; Sun, H.; Costa, M. Molecular and epigenetic mechanisms of Cr(VI)-induced carcinogenesis. Toxicol. Appl. Pharmacol. 2019, 377, 114636. [Google Scholar] [CrossRef]
- Chen, B.; Xiong, J.; Ding, J.H.; Yuan, B.F.; Feng, Y.Q. Analysis of the Effects of Cr(VI) Exposure on mRNA Modifications. Chem. Res. Toxicol. 2019, 32, 2078–2085. [Google Scholar] [CrossRef]
- Lv, Y.; Li, T.; Yang, M.; Su, L.; Zhu, Z.; Zhao, S.; Zeng, W.; Zheng, Y. Melatonin Attenuates Chromium (VI)-Induced Spermatogonial Stem Cell/Progenitor Mitophagy by Restoration of METTL3-Mediated RNA N 6-Methyladenosine Modification. Front. Cell. Dev. Biol. 2021, 9, 684398. [Google Scholar] [CrossRef]
- Wang, Z.; Uddin, M.B.; Xie, J.; Tao, H.; Zeidler-Erdely, P.C.; Kondo, K.; Yang, C. Chronic Hexavalent Chromium Exposure Up-regulates the RNA Methyltransferase METTL3 Expression to Promote Cell Transformation; Cancer Stem Cell-like Property and Tumorigenesis. Toxicol. Sci. 2022, 187, 51–61. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Wang, Z. The Epitranscriptomic Mechanism of Metal Toxicity and Carcinogenesis. Int. J. Mol. Sci. 2022, 23, 11830. https://doi.org/10.3390/ijms231911830
Yang C, Wang Z. The Epitranscriptomic Mechanism of Metal Toxicity and Carcinogenesis. International Journal of Molecular Sciences. 2022; 23(19):11830. https://doi.org/10.3390/ijms231911830
Chicago/Turabian StyleYang, Chengfeng, and Zhishan Wang. 2022. "The Epitranscriptomic Mechanism of Metal Toxicity and Carcinogenesis" International Journal of Molecular Sciences 23, no. 19: 11830. https://doi.org/10.3390/ijms231911830
APA StyleYang, C., & Wang, Z. (2022). The Epitranscriptomic Mechanism of Metal Toxicity and Carcinogenesis. International Journal of Molecular Sciences, 23(19), 11830. https://doi.org/10.3390/ijms231911830