
Structural and Electronic Effects at the Interface between Transition Metal Dichalcogenide Monolayers (MoS2, WSe2, and Their Lateral Heterojunctions) and Liquid Water


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussions
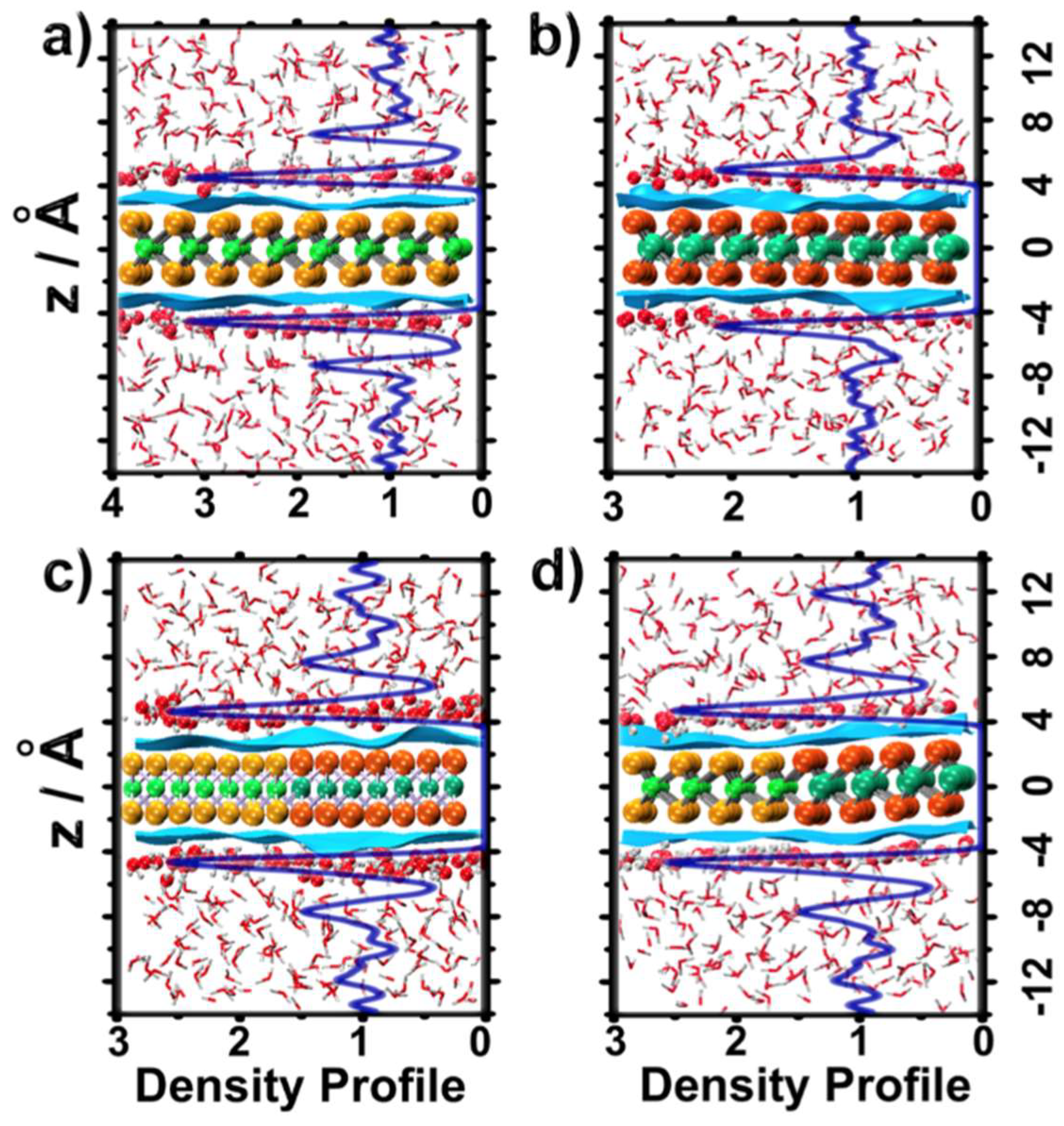
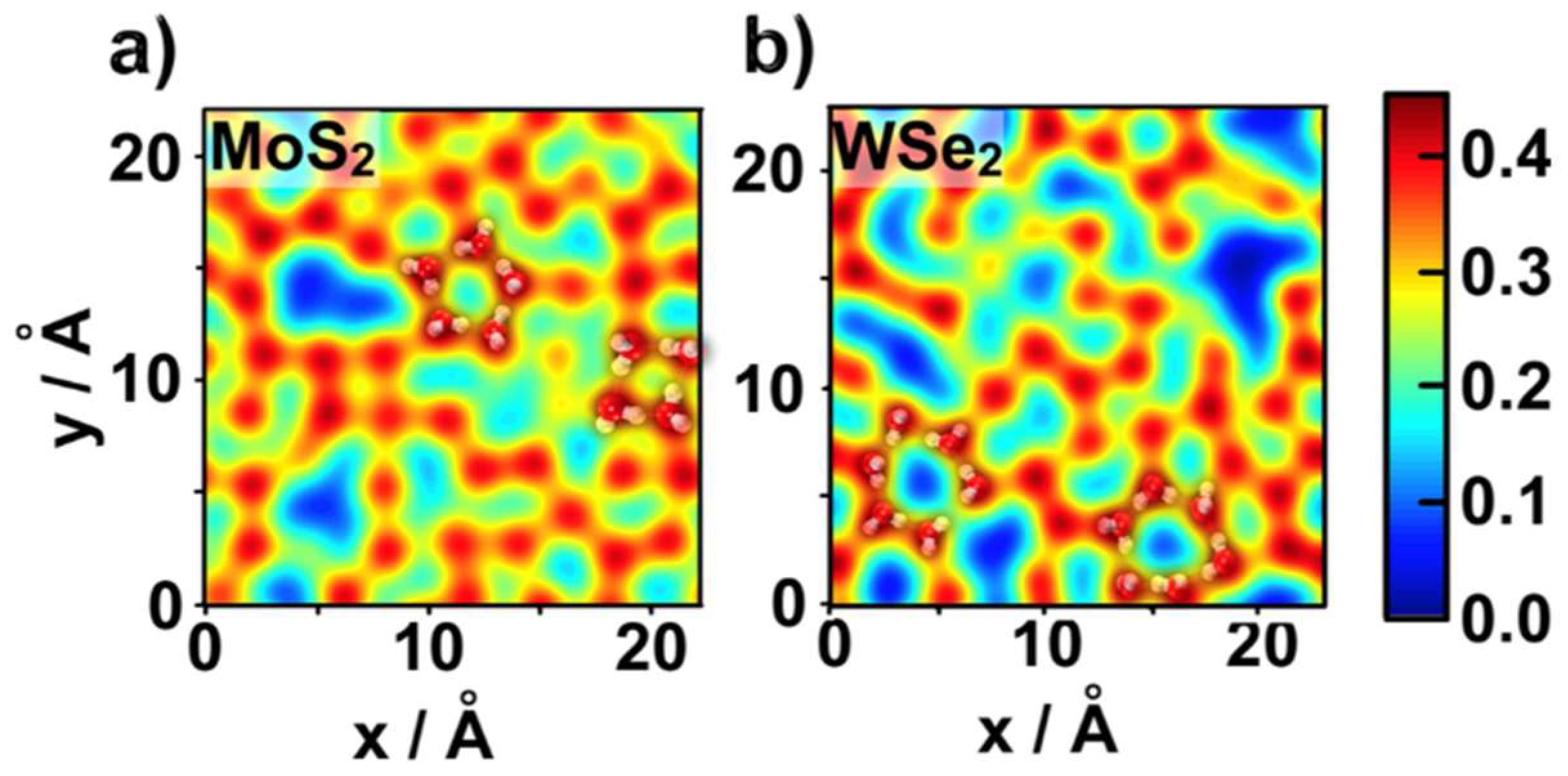
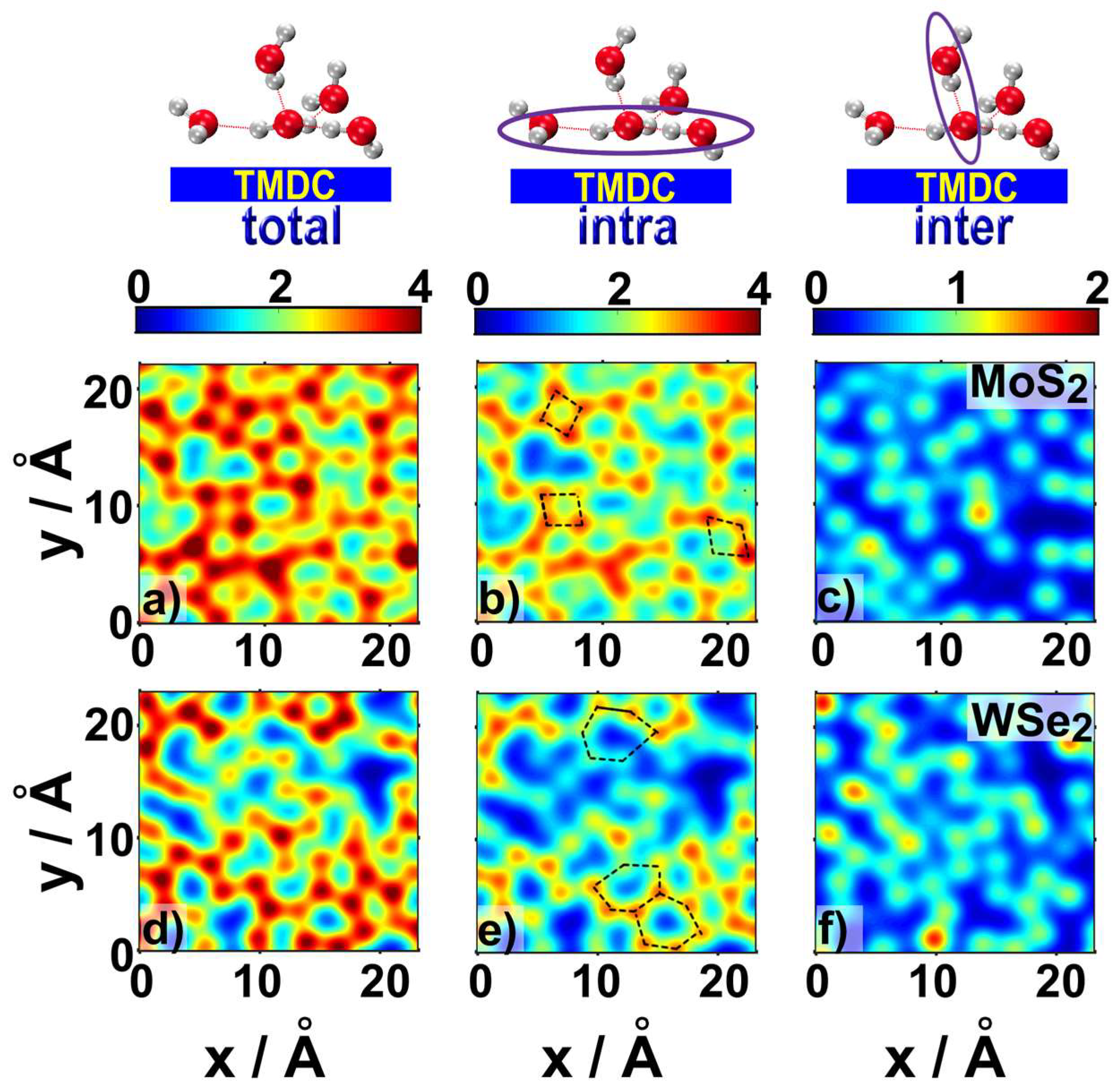
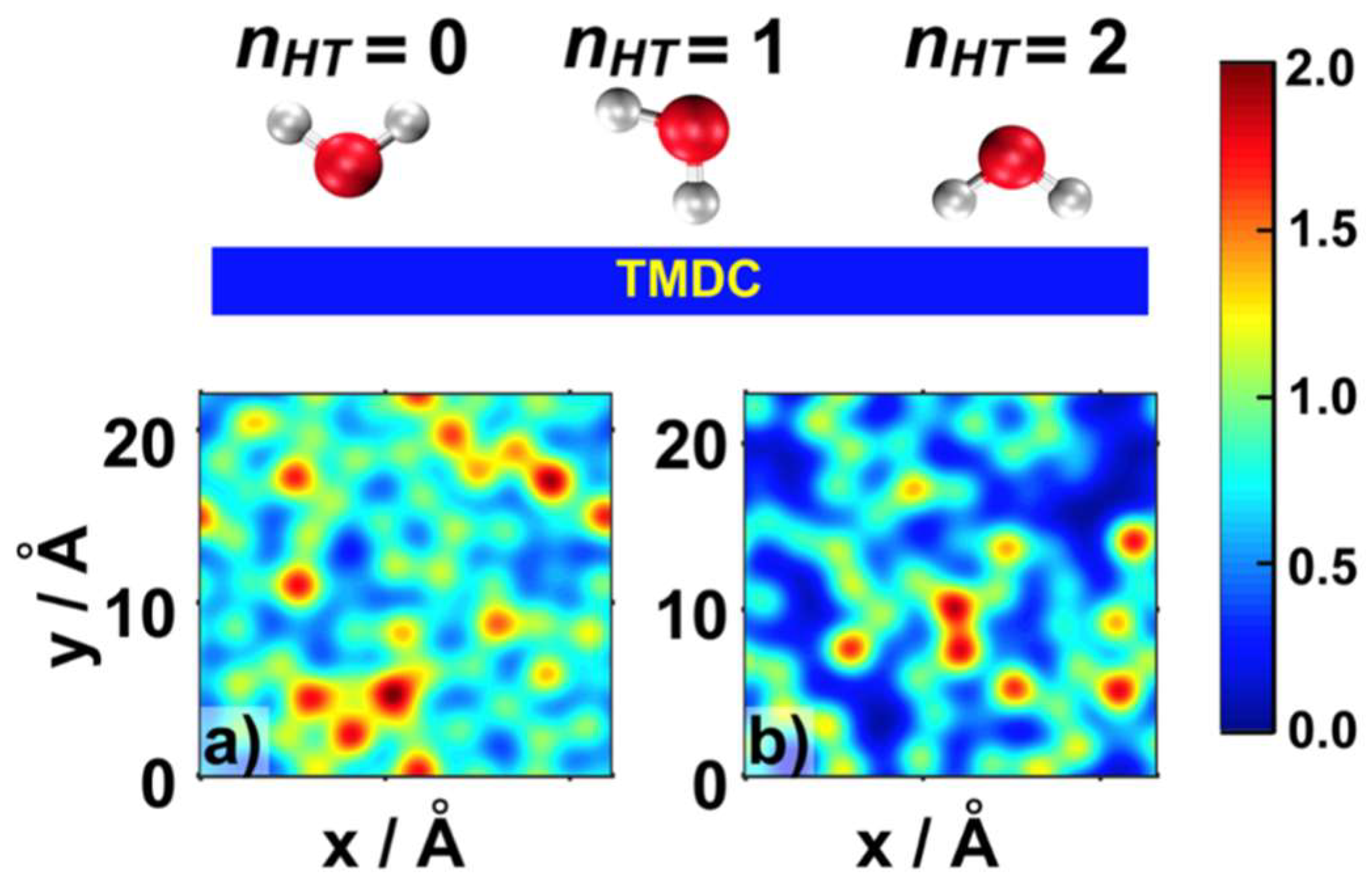
2.1. Water Structure at the TMDCs-Water Interface
2.2. Electronic Properties of Wetted TMDCs
3. Simulation Methods
3.1. Empirical Force Field Development
3.2. Classical Molecular Dynamics Simulations
3.3. Density Functional Theory Molecular Dynamics (DFT-MD) Simulations
3.4. Density Functional Theory Geometry Optimizations
3.5. Electronic Property Calculations
4. Conclusions
- Water molecules adsorbed on TMDCs surfaces formed high-density water layers, with differently structured patterns of H-bonded water molecules (~4 H-bonds per water molecule) on different TMDC materials. These patterns were indicative of the hydrophobic character of the TMDC surface;
- Some water molecules in the ad-layer were connected to bulk water through H-bonds, while some other waters had hydrogen atoms oriented towards the TMDC surface. This could facilitate proton transfer from bulk water to the TMDC surface via the Grotthuss mechanism in electrochemical or photocatalytic applications;
- Despite the general trends described above, the ad-layers on top of the two TMDCs examined were different. The topology on top of MoS2 was denser, with a structure presenting four to six-water rings; while the topology on top of WSe2 had a higher tendency to form six-water rings;
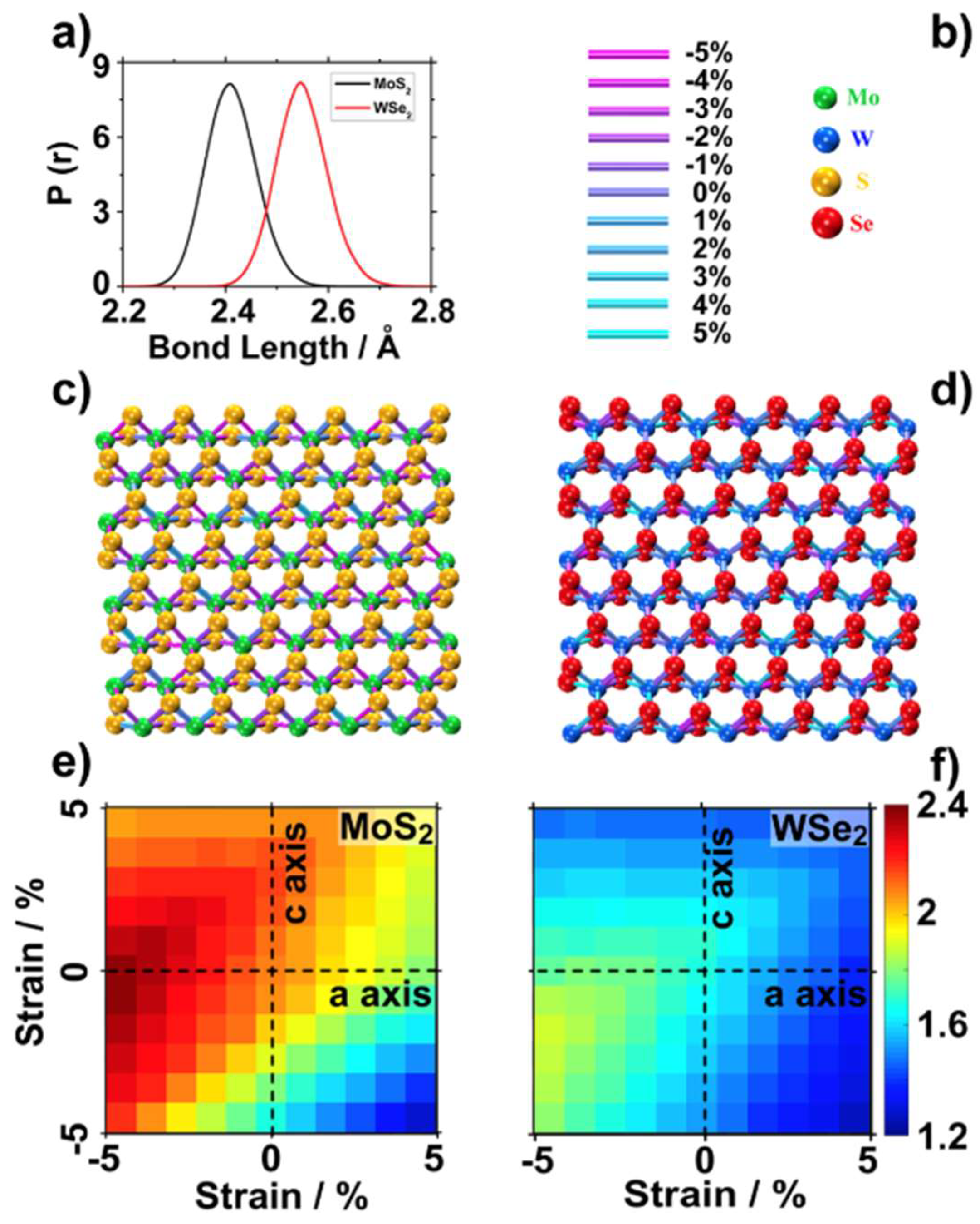
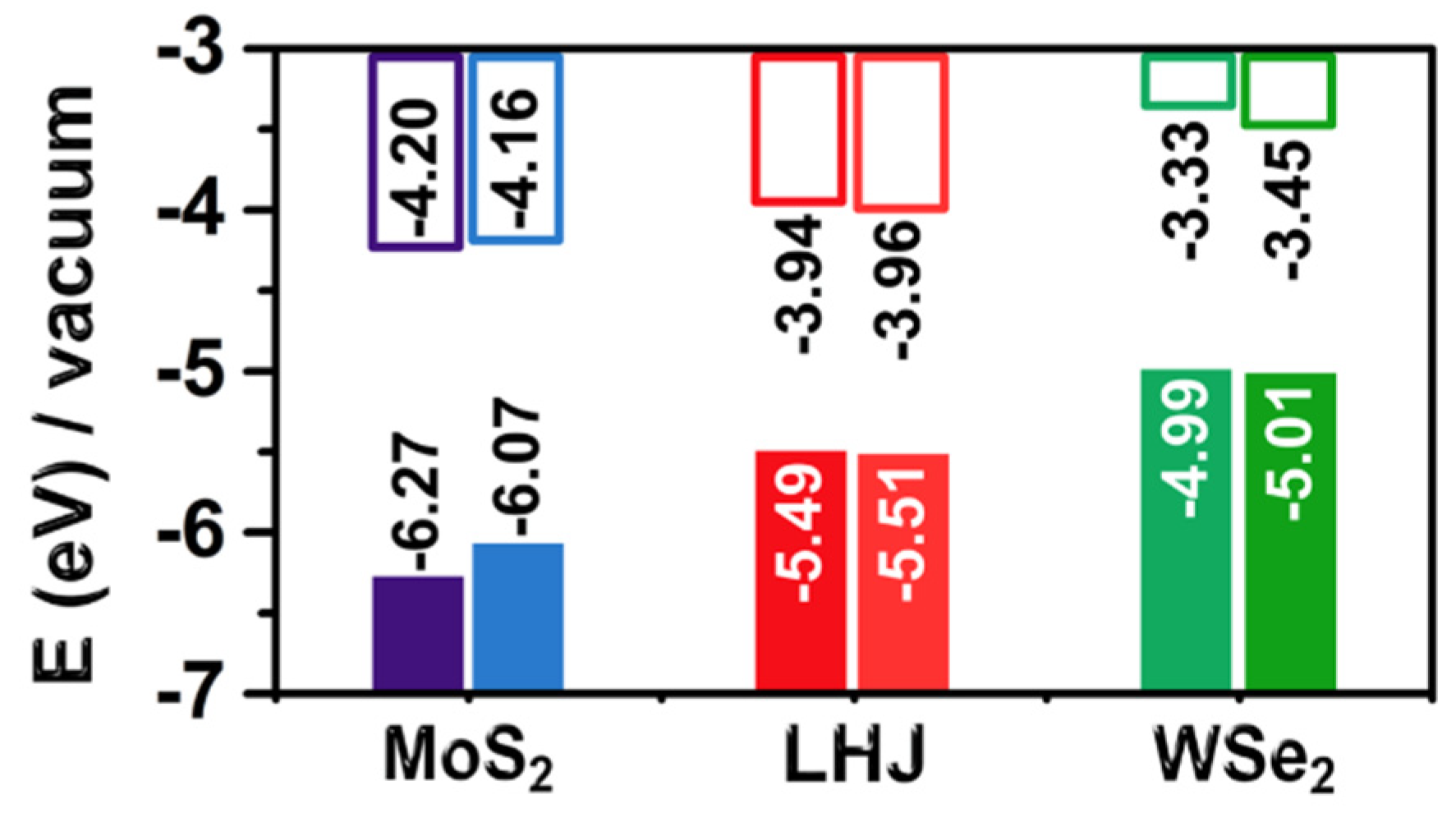
- The metal chalcogenide bonds of the TMDCs fluctuate up to ±5% from the equilibrium value during dynamics. Due to the anisotropic band gap dependence on deformations, the effects of stochastic deformation during the DFT-MD simulations tend to compensate each other. As a result, the band gap of MoS2 decreased by up to 0.2 eV. A smaller impact, 0.1 eV, was found on the band gap of WSe2. The impact of the TMDC structural deformations on the band alignment to vacuum was minimal;
- The presence of explicit water molecules had similar negligible effect, below 0.05 eV, on the band gap and band alignment of the TMDCs. This result was consistent with the hydrophobic character and the apolar nature of infinite TMDC monolayers. Analysis of frames from the DFT-MD simulations evidenced a small effect of water and dynamics on the light absorption spectra, limited to a somewhat weaker absorption at shorter wavelengths and formation of a shoulder at longer wavelengths.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, Q.; Song, B.; Xu, P.; Jin, S. Efficient Electrocatalytic and Photoelectrochemical Hydrogen Generation Using MoS2 and Related Compounds. Chem 2016, 1, 699–726. [Google Scholar] [CrossRef]
- Faber, M.S.; Jin, S. Earth-abundant inorganic electrocatalysts and their nanostructures for energy conversion applications. Energy Environ. Sci. 2014, 7, 3519–3542. [Google Scholar] [CrossRef]
- Ganguly, P.; Harb, M.; Cao, Z.; Cavallo, L.; Breen, A.; Dervin, S.; Dionysiou, D.D.; Pillai, S.C. 2D Nanomaterials for Photocatalytic Hydrogen Production. ACS Energy Lett. 2019, 4, 1687–1709. [Google Scholar] [CrossRef]
- Laursen, A.B.; Kegnæs, S.; Dahl, S.; Chorkendorff, I. Molybdenum sulfides—Efficient and viable materials for electro- and photoelectrocatalytic hydrogen evolution. Energy Environ. Sci. 2012, 5, 5577–5591. [Google Scholar] [CrossRef]
- Vesborg, P.C.K.; Seger, B.; Chorkendorff, I. Recent Development in Hydrogen Evolution Reaction Catalysts and Their Practical Implementation. J. Phys. Chem. Lett. 2015, 6, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, B.; Wang, H.; Li, Y.; Zuo, Y.; Wang, W.; Lin, H.; Li, S.; Wang, L. Efficient spatial charge separation in unique 2D tandem heterojunction CdxZn1−xIn2S4–CdS–MoS2 rendering highly-promoted visible-light-induced H2 generation. J. Mater. Chem. A 2021, 9, 482–491. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, K.; Ma, C.; Chen, Y.; Zhang, H.; Liu, Y.; Ji, Y. Novel two-dimensional β-GeSe and β-SnSe semiconductors: Anisotropic high carrier mobility and excellent photocatalytic water splitting. J. Mater. Chem. A 2020, 8, 19612–19622. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Zavabeti, A.; Haque, F.; Mahmood, A.; Datta, R.S.; Syed, N.; Khan, M.W.; Jannat, A.; Messalea, K.; Zhang, B.Y.; et al. Synthesis of two-dimensional hematite and iron phosphide for hydrogen evolution. J. Mater. Chem. A 2020, 8, 2789–2797. [Google Scholar] [CrossRef]
- Zheng, B.; Chen, Y.; Qi, F.; Wang, X.; Zhang, W.; Li, Y.; Li, X. 3D-hierarchical MoSe 2 nanoarchitecture as a highly efficient electrocatalyst for hydrogen evolution. 2D Mater. 2017, 4, 025092. [Google Scholar] [CrossRef]
- Roeb, M.; Monnerie, N.; Houaijia, A.; Thomey, D.; Sattler, C. Chapter 4—Solar Thermal Water Splitting. In Renewable Hydrogen Technologies; Gandía, L.M., Arzamendi, G., Diéguez, P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 63–86. [Google Scholar]
- Fu, Q.; Han, J.; Wang, X.; Xu, P.; Yao, T.; Zhong, J.; Zhong, W.; Liu, S.; Gao, T.; Zhang, Z.; et al. 2D Transition Metal Dichalcogenides: Design, Modulation, and Challenges in Electrocatalysis. Adv. Mater. 2021, 33, 1907818. [Google Scholar] [CrossRef]
- Kadam, S.R.; Enyashin, A.N.; Houben, L.; Bar-Ziv, R.; Bar-Sadan, M. Ni–WSe2 nanostructures as efficient catalysts for electrochemical hydrogen evolution reaction (HER) in acidic and alkaline media. J. Mater. Chem. A 2020, 8, 1403–1416. [Google Scholar] [CrossRef]
- Mazánek, V.; Mayorga-Martinez, C.C.; Bouša, D.; Sofer, Z.; Pumera, M. WSe2 nanoparticles with enhanced hydrogen evolution reaction prepared by bipolar electrochemistry: Application in competitive magneto-immunoassay. Nanoscale 2018, 10, 23149–23156. [Google Scholar] [CrossRef]
- Lee, J.; Heo, J.; Lim, H.Y.; Seo, J.; Kim, Y.; Kim, J.; Kim, U.; Choi, Y.; Kim, S.H.; Yoon, Y.J.; et al. Defect-Induced in Situ Atomic Doping in Transition Metal Dichalcogenides via Liquid-Phase Synthesis toward Efficient Electrochemical Activity. ACS Nano 2020, 14, 17114–17124. [Google Scholar] [CrossRef]
- Chia, H.L.; Mayorga-Martinez, C.C.; Sofer, Z.; Lazar, P.; Webster, R.D.; Pumera, M. Vanadium Dopants: A Boon or a Bane for Molybdenum Dichalcogenides-Based Electrocatalysis Applications. Adv. Funct. Mater. 2021, 31, 2009083. [Google Scholar] [CrossRef]
- Sulas-Kern, D.B.; Miller, E.M.; Blackburn, J.L. Photoinduced charge transfer in transition metal dichalcogenide heterojunctions—Towards next generation energy technologies. Energy Environ. Sci. 2020, 13, 2684–2740. [Google Scholar] [CrossRef]
- Guo, S.; Li, Y.; Tang, S.; Zhang, Y.; Li, X.; Sobrido, A.J.; Titirici, M.-M.; Wei, B. Monitoring Hydrogen Evolution Reaction Intermediates of Transition Metal Dichalcogenides via Operando Raman Spectroscopy. Adv. Funct. Mater. 2020, 30, 2003035. [Google Scholar] [CrossRef]
- Zheng, Z.; Yu, L.; Gao, M.; Chen, X.; Zhou, W.; Ma, C.; Wu, L.; Zhu, J.; Meng, X.; Hu, J.; et al. Boosting hydrogen evolution on MoS2 via co-confining selenium in surface and cobalt in inner layer. Nat. Commun. 2020, 11, 3315. [Google Scholar] [CrossRef]
- Li, H.; Du, M.; Mleczko, M.J.; Koh, A.L.; Nishi, Y.; Pop, E.; Bard, A.J.; Zheng, X. Kinetic Study of Hydrogen Evolution Reaction over Strained MoS2 with Sulfur Vacancies Using Scanning Electrochemical Microscopy. J. Am. Chem. Soc. 2016, 138, 5123–5129. [Google Scholar] [CrossRef]
- Huang, Y.; Nielsen, R.J.; Goddard, W.A. Reaction Mechanism for the Hydrogen Evolution Reaction on the Basal Plane Sulfur Vacancy Site of MoS2 Using Grand Canonical Potential Kinetics. J. Am. Chem. Soc. 2018, 140, 16773–16782. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, H.; Guo, C.; Cao, S.; Wu, C.-M.L.; Wang, Z.; Lu, X. Theoretical investigation on the hydrogen evolution reaction mechanism at MoS2 heterostructures: The essential role of the 1T/2H phase interface. Catal. Sci. Technol. 2020, 10, 458–465. [Google Scholar] [CrossRef]
- Wang, N.; Cao, Z.; Zheng, X.; Zhang, B.; Kozlov, S.M.; Chen, P.; Zou, C.; Kong, X.; Wen, Y.; Liu, M.; et al. Hydration-Effect-Promoting Ni–Fe Oxyhydroxide Catalysts for Neutral Water Oxidation. Adv. Mater. 2020, 32, 1906806. [Google Scholar] [CrossRef]
- Shinagawa, T.; Takanabe, K. New Insight into the Hydrogen Evolution Reaction under Buffered Near-Neutral pH Conditions: Enthalpy and Entropy of Activation. J. Phys. Chem. C 2016, 120, 24187–24196. [Google Scholar] [CrossRef]
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.-M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef]
- Bellarosa, L.; García-Muelas, R.; Revilla-López, G.; López, N. Diversity at the Water–Metal Interface: Metal, Water Thickness, and Confinement Effects. ACS Cent. Sci. 2016, 2, 109–116. [Google Scholar] [CrossRef]
- Cao, Z.; Kumar, R.; Peng, Y.; Voth, G.A. Proton Transport under External Applied Voltage. J. Phys. Chem. B 2014, 118, 8090–8098. [Google Scholar] [CrossRef]
- Limmer, D.T.; Willard, A.P.; Madden, P.; Chandler, D. Hydration of metal surfaces can be dynamically heterogeneous and hydrophobic. Proc. Natl. Acad. Sci. USA 2013, 110, 4200–4205. [Google Scholar] [CrossRef]
- Pham, T.A.; Ping, Y.; Galli, G. Modelling heterogeneous interfaces for solar water splitting. Nat. Mater. 2017, 16, 401–408. [Google Scholar] [CrossRef]
- Pham, T.A.; Lee, D.; Schwegler, E.; Galli, G. Interfacial Effects on the Band Edges of Functionalized Si Surfaces in Liquid Water. J. Am. Chem. Soc. 2014, 136, 17071–17077. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Kumar, R.; Peng, Y.; Voth, G.A. Hydrated Proton Structure and Diffusion at Platinum Surfaces. J. Phys. Chem. C 2015, 119, 14675–14682. [Google Scholar] [CrossRef]
- Gaur, A.P.S.; Sahoo, S.; Ahmadi, M.; Dash, S.P.; Guinel, M.J.F.; Katiyar, R.S. Surface Energy Engineering for Tunable Wettability through Controlled Synthesis of MoS2. Nano Lett. 2014, 14, 4314–4321. [Google Scholar] [CrossRef] [PubMed]
- Heyne, M.H.; Chiappe, D.; Meersschaut, J.; Nuytten, T.; Conard, T.; Bender, H.; Huyghebaert, C.; Radu, I.P.; Caymax, M.; de Marneffe, J.F.; et al. Multilayer MoS2 growth by metal and metal oxide sulfurization. J. Mater. Chem. C 2016, 4, 1295–1304. [Google Scholar] [CrossRef]
- Carrasco, J.; Hodgson, A.; Michaelides, A. A molecular perspective of water at metal interfaces. Nat. Mater. 2012, 11, 667–674. [Google Scholar] [CrossRef]
- Duncan, W.R.; Prezhdo, O.V. Theoretical Studies of Photoinduced Electron Transfer in Dye-Sensitized TiO2. Ann. Rev. Phys. Chem. 2007, 58, 143–184. [Google Scholar] [CrossRef]
- Willard, A.P.; Chandler, D. Instantaneous Liquid Interfaces. J. Phys. Chem. B 2010, 114, 1954–1958. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Willard, A.P.; Chandler, D. The molecular structure of the interface between water and a hydrophobic substrate is liquid-vapor like. J. Chem. Phys. 2014, 141, 18C519. [Google Scholar] [CrossRef]
- Boroda, Y.G.; Calhoun, A.; Voth, G.A. A theory for electron transfer across the electrode/electrolyte interface involving more than one redox ion. J. Chem. Phys. 1997, 107, 8940–8954. [Google Scholar] [CrossRef]
- Gallo, P.; Amann-Winkel, K.; Angell, C.A.; Anisimov, M.A.; Caupin, F.; Chakravarty, C.; Lascaris, E.; Loerting, T.; Panagiotopoulos, A.Z.; Russo, J.; et al. Water: A Tale of Two Liquids. Chem. Rev. 2016, 116, 7463–7500. [Google Scholar] [CrossRef]
- Merte, L.R.; Peng, G.; Bechstein, R.; Rieboldt, F.; Farberow, C.A.; Grabow, L.C.; Kudernatsch, W.; Wendt, S.; Lægsgaard, E.; Mavrikakis, M.; et al. Water-Mediated Proton Hopping on an Iron Oxide Surface. Science 2012, 336, 889–893. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Jain, A.; de Arquer, F.P.G.; De Luna, P.; Li, J.; Wang, N.; Zheng, X.; Cai, J.; Gregory, B.Z.; Voznyy, O.; et al. Multi-site electrocatalysts for hydrogen evolution in neutral media by destabilization of water molecules. Nat. Energy 2019, 4, 107–114. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Zhu, Y.; Li, L.H.; Han, Y.; Chen, Y.; Jaroniec, M.; Qiao, S.-Z. High Electrocatalytic Hydrogen Evolution Activity of an Anomalous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 16174–16181. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Chen, Y.; Zhang, W.; Quek, S.Y.; Chen, C.-H.; Li, L.-J.; Hsu, W.-T.; Chang, W.-H.; Zheng, Y.J.; Chen, W.; et al. Bandgap tunability at single-layer molybdenum disulphide grain boundaries. Nat. Commun. 2015, 6, 6298. [Google Scholar] [CrossRef] [PubMed]
- Chaves, A.; Azadani, J.G.; Alsalman, H.; da Costa, D.R.; Frisenda, R.; Chaves, A.J.; Song, S.H.; Kim, Y.D.; He, D.; Zhou, J.; et al. Bandgap engineering of two-dimensional semiconductor materials. NPJ 2D Mater. Appl. 2020, 4, 29. [Google Scholar] [CrossRef]
- Cao, Z.; Harb, M.; Lardhi, S.; Cavallo, L. Impact of Interfacial Defects on the Properties of Monolayer Transition Metal Dichalcogenide Lateral Heterojunctions. J. Phys. Chem. Lett. 2017, 8, 1664–1669. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Steiner, S.; Khmelevskyi, S.; Marsmann, M.; Kresse, G. Calculation of the magnetic anisotropy with projected-augmented-wave methodology and the case study of disordered Fe1−xCox alloys. Phys. Rev. B 2016, 93, 224425. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Harb, M.; Kozlov, S.M.; Cavallo, L. Structural and Electronic Effects at the Interface between Transition Metal Dichalcogenide Monolayers (MoS2, WSe2, and Their Lateral Heterojunctions) and Liquid Water. Int. J. Mol. Sci. 2022, 23, 11926. https://doi.org/10.3390/ijms231911926
Cao Z, Harb M, Kozlov SM, Cavallo L. Structural and Electronic Effects at the Interface between Transition Metal Dichalcogenide Monolayers (MoS2, WSe2, and Their Lateral Heterojunctions) and Liquid Water. International Journal of Molecular Sciences. 2022; 23(19):11926. https://doi.org/10.3390/ijms231911926
Chicago/Turabian StyleCao, Zhen, Moussab Harb, Sergey M. Kozlov, and Luigi Cavallo. 2022. "Structural and Electronic Effects at the Interface between Transition Metal Dichalcogenide Monolayers (MoS2, WSe2, and Their Lateral Heterojunctions) and Liquid Water" International Journal of Molecular Sciences 23, no. 19: 11926. https://doi.org/10.3390/ijms231911926
APA StyleCao, Z., Harb, M., Kozlov, S. M., & Cavallo, L. (2022). Structural and Electronic Effects at the Interface between Transition Metal Dichalcogenide Monolayers (MoS2, WSe2, and Their Lateral Heterojunctions) and Liquid Water. International Journal of Molecular Sciences, 23(19), 11926. https://doi.org/10.3390/ijms231911926