
The Defects of Epigenetic Reprogramming in Dox-Dependent Porcine-iPSCs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
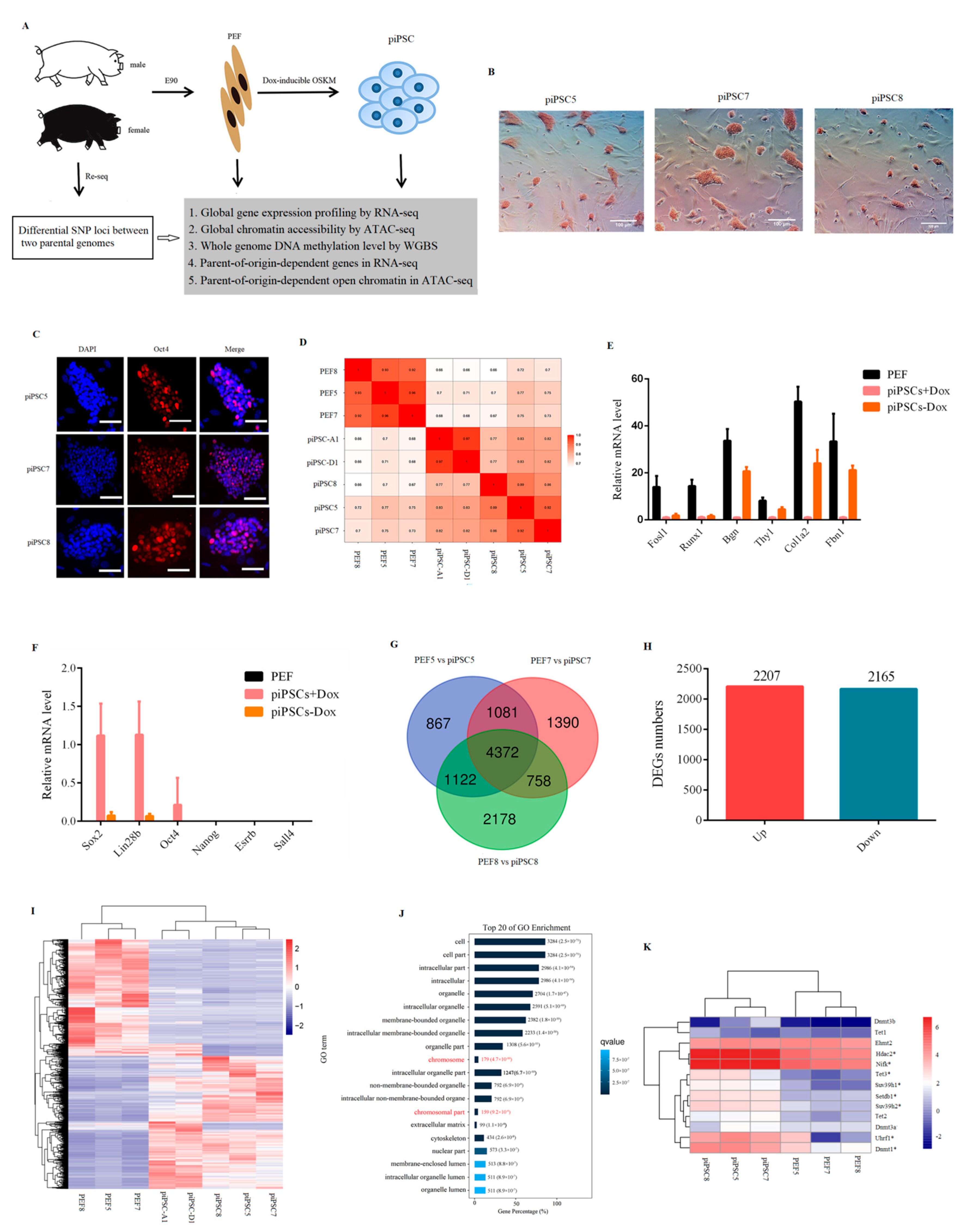
2.1. Generation of piPSCs Genetically Matched with PEF
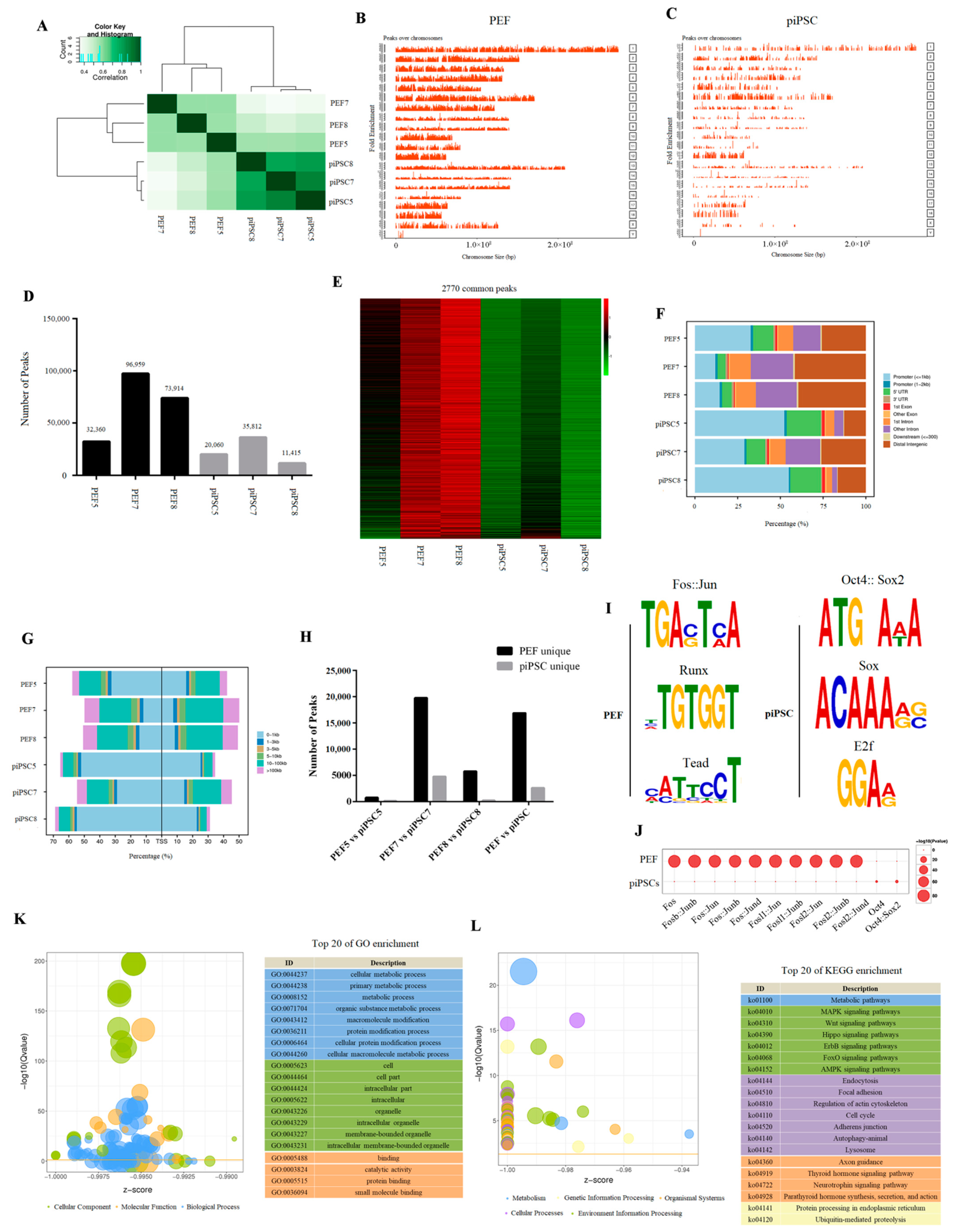
2.2. Accessible Chromatin Was Changed after Reprogramming
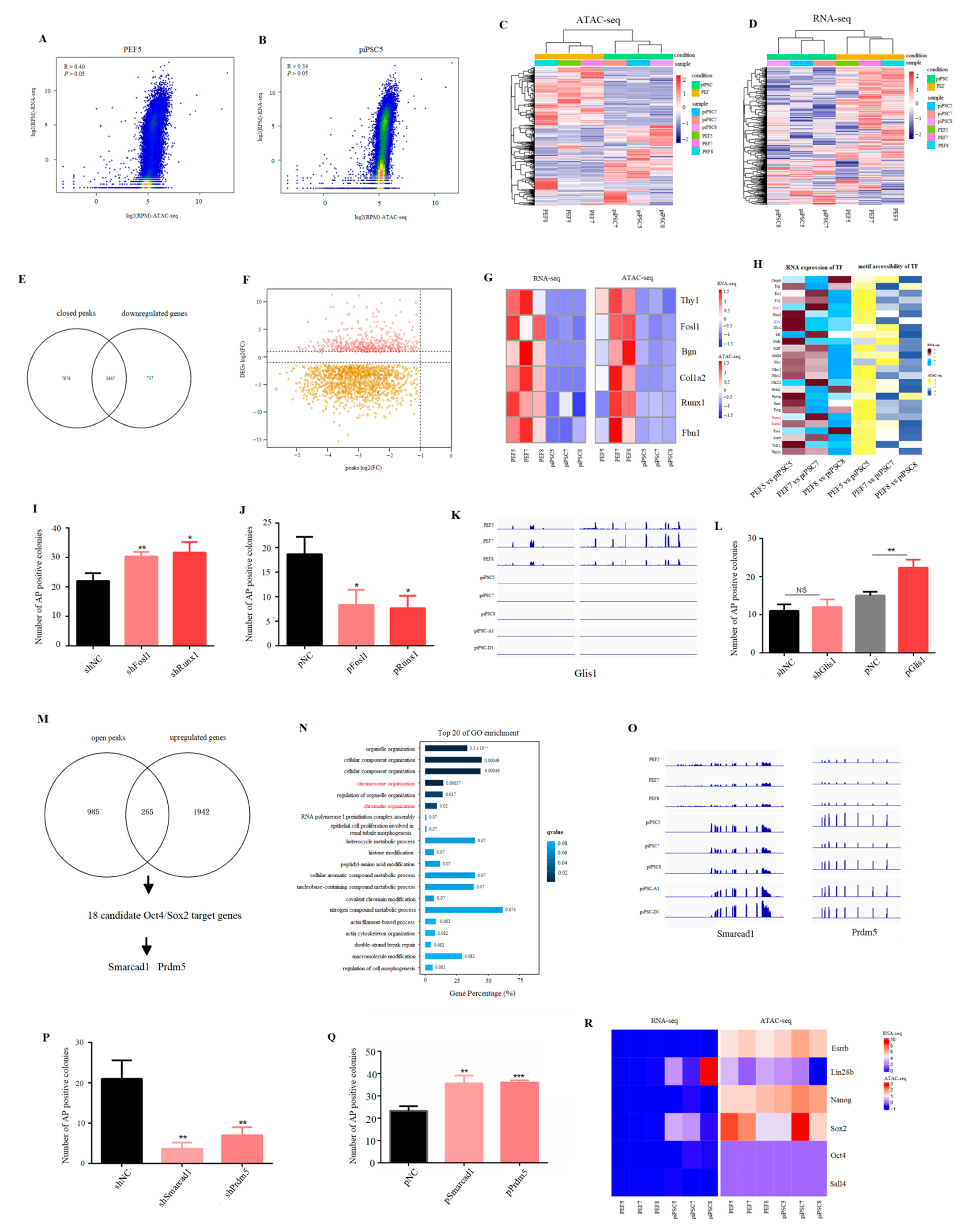
2.3. Effect of Accessible Chromatin on Gene Expression
2.4. Loss of Global DNA Methylation after piPSC Reprogramming
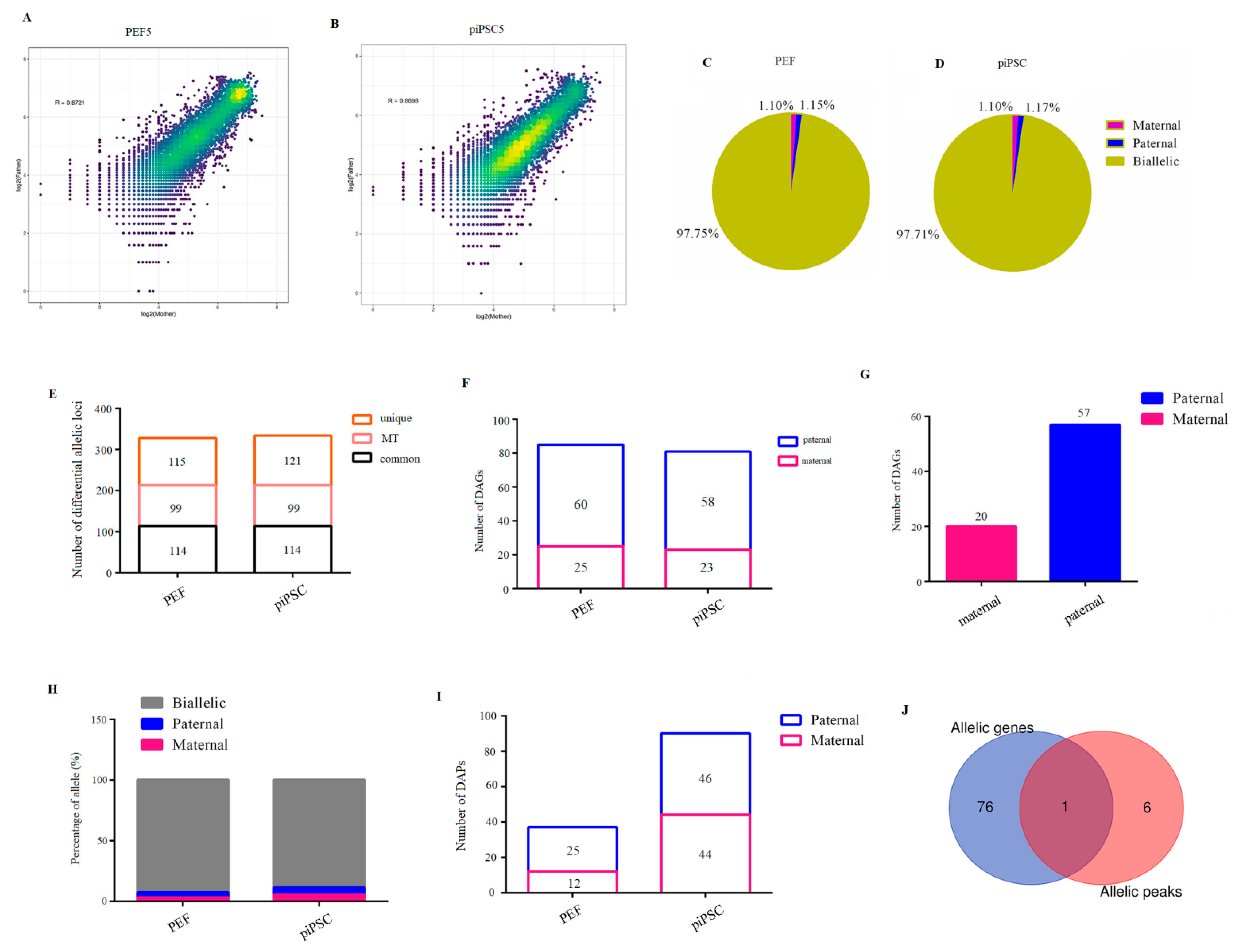
2.5. Identification of Allele-Biased Expressed Genes and Allele-Biased Open Chromatin
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Generation of piPSCs
4.4. Alkaline Phosphatase (AP) Staining
4.5. Immunofluorescence Assay
4.6. Library Construction and Sequencing
4.7. Identification of Parent-of-Origin-Dependent Genes
4.8. Identification of Parent-of-Origin-Dependent Accessible Chromatin Regions
4.9. RNA Oligonucleotides, Cell Transfection, and Real-Time Quantitative PCR
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waddington, C.H. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology; With an Appendix by H. Kacser; George Allen & Unwin: London, UK, 1957. [Google Scholar]
- Teramoto, Y.; Takahashi, D.Y.; Holmes, P.; A Ghazanfar, A. Vocal development in a Waddington landscape. eLife 2017, 6. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into iPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Hussein, S.M.I.; Puri, M.C.; Tonge, P.D.; Benevento, M.; Corso, A.J.; Clancy, J.L.; Mosbergen, R.; Li, M.; Lee, D.-S.; Cloonan, N.; et al. Genome-wide characterization of the routes to pluripotency. Nature 2014, 516, 198–206. [Google Scholar] [CrossRef]
- Buganim, Y.; Markoulaki, S.; van Wietmarschen, N.; Hoke, H.; Wu, T.; Ganz, K.; Akhtar-Zaidi, B.; He, Y.; Abraham, B.J.; Porubsky, D.; et al. The Developmental Potential of iPSCs Is Greatly Influenced by Reprogramming Factor Selection. Cell Stem Cell 2014, 15, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koche, R.P.; Smith, Z.D.; Adli, M.; Gu, H.; Ku, M.; Gnirke, A.; Bernstein, B.E.; Meissner, A. Reprogramming Factor Expression Initiates Widespread Targeted Chromatin Remodeling. Cell Stem Cell 2011, 8, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, J.; Yang, X.; Zhou, C.; Guo, J.; Wu, C.; Qin, Y.; Guo, L.; He, J.; Yu, S.; et al. Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell 2017, 21, 819–833.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chronis, C.; Fiziev, P.; Papp, B.; Butz, S.; Bonora, G.; Sabri, S.; Ernst, J.; Plath, K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 2017, 168, 442–459.e20. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, D.D.; You, J.S.; Jones, P.A. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010, 20, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Long, W.; Zhang, X.; Xu, K.; Guo, J.; Zhao, H.; Li, H.; Qing, Y.; Pan, W.; Jia, B.; et al. Generation of GHR-modified pigs as Laron syndrome models via a dual-sgRNAs/Cas9 system and somatic cell nuclear transfer. J. Transl. Med. 2018, 16, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Chen, J.; Ren, J.; Bao, L.; Liao, J.; Cui, C.; Rao, L.; Li, H.; Gu, Y.; Dai, H.; et al. Generation of Pig Induced Pluripotent Stem Cells with a Drug-Inducible System. J. Mol. Cell Biol. 2009, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Nowak-Imialek, M.; Chen, X.; Chen, D.; Herrmann, D.; Ruan, D.; Chen, A.C.H.; Eckersley-Maslin, M.A.; Ahmad, S.; Lee, Y.L.; et al. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 2019, 21, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, S.C.; Surani, M.A.H.; Norris, M.L. Role of paternal and maternal genomes in mouse development. Nature 1984, 311, 374–376. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Henckel, A.; Arnaud, P. Genome-wide identification of new imprinted genes. Briefings Funct. Genom. 2010, 9, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Clark, A.G. Using next-generation RNA sequencing to identify imprinted genes. Heredity 2014, 113, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Dai, M.; Li, F.; Liu, A. Genomic imprinting, methylation and parent-of-origin effects in reciprocal hybrid endosperm of castor bean. Nucleic Acids Res. 2014, 42, 6987–6998. [Google Scholar] [CrossRef]
- DeVeale, B.; Van Der Kooy, D.; Babak, T. Critical Evaluation of Imprinted Gene Expression by RNA–Seq: A New Perspective. PLoS Genet. 2012, 8, e1002600. [Google Scholar] [CrossRef] [PubMed]
- Okae, H.; Hiura, H.; Nishida, Y.; Funayama, R.; Tanaka, S.; Chiba, H.; Yaegashi, N.; Nakayama, K.; Sasaki, H.; Arima, T. Re-investigation and RNA sequencing-based identification of genes with placenta-specific imprinted expression. Hum. Mol. Genet. 2011, 21, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-E.; Luo, A.; Xin, H.-P.; Zhao, J.; Li, S.-S.; Qu, L.-H.; Ma, L.-G.; Scholten, S.; Sun, M.-X. Genes of Both Parental Origins Are Differentially Involved in Early Embryogenesis of a Tobacco Interspecies Hybrid. PLoS ONE 2011, 6, e23153. [Google Scholar] [CrossRef] [PubMed]
- DeVeale, B.; van der Kooy, D. Parental Bias Has Benefits. Neuron 2020, 107, 994–996. [Google Scholar] [CrossRef]
- Liu, X.; Nefzger, C.; Rossello, F.J.; Chen, J.; Knaupp, A.; Firas, J.; Ford, E.; Pflueger, J.; Paynter, J.M.; Chy, H.S.; et al. Comprehensive characterization of distinct states of human naive pluripotency generated by reprogramming. Nat. Methods 2017, 14, 1055–1062. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, T.; Cai, Y.; Wang, H. Preserving self-renewal of porcine pluripotent stem cells in serum-free 3i culture condition and independent of LIF and b-FGF cytokines. Cell Death Discov. 2018, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 facilitates induction of pluripotency via an epigenome–metabolome–epigenome signalling cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Sharov, A.A.; Masui, S.; Sharova, L.V.; Piao, Y.; Aiba, K.; Matoba, R.; Xin, L.; Niwa, H.; Ko, M.S. Identification of Pou5f1, Sox2, and Nanog downstream target genes with statistical confidence by applying a novel algorithm to time course microarray and genome-wide chromatin immunoprecipitation data. BMC Genomics. 2008, 9, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef] [PubMed]
- Tchieu, J.; Kuoy, E.; Chin, M.H.; Trinh, H.; Patterson, M.; Sherman, S.P.; Aimiuwu, O.; Lindgren, A.; Hakimian, S.; Zack, J.A.; et al. Female Human iPSCs Retain an Inactive X Chromosome. Cell Stem Cell 2010, 7, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar] [CrossRef]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Li, D.; Liu, Y.; Guo, J.; Li, C.; Yao, Y.; Wang, Y.; Zhao, G.; Wang, X.; et al. Induction of Pluripotent Stem Cells from Mouse Embryonic Fibroblasts by Jdp2-Jhdm1b-Mkk6-Glis1-Nanog-Essrb-Sall4. Cell Rep. 2019, 27, 3473–3485.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.G.; Frampton, G.M.; Soldner, F.; Hockemeyer, D.; Mitalipova, M.; Jaenisch, R.; Young, R.A. Chromatin Structure and Gene Expression Programs of Human Embryonic and Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veazey, K.J.; Wang, H.; Bedi, Y.S.; Skiles, W.M.; Chang, R.C.; Golding, M.C. Disconnect between alcohol-induced alterations in chromatin structure and gene transcription in a mouse embryonic stem cell model of exposure. Alcohol 2017, 60, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Barlow, D.P.; Bartolomei, M. Genomic Imprinting in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Wu, J.; Huang, B.; Chen, H.; Yin, Q.; Liu, Y.; Xiang, Y.; Zhang, B.; Liu, B.; Wang, Q.; Xia, W.; et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016, 534, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Yin, D.; Zhang, L.; Li, B.; Li, R.; Zhang, X.; Zhang, Z.; Liu, H.; Kim, K.; Wu, W. Parsing the microRNA genetics basis regulating skeletal muscle fiber types and meat quality traits in pigs. Anim. Genet. 2021, 52, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Jiang, A.; Guo, H.; Wu, W.; Liu, H. The Crosstalk between Autophagy and Apoptosis Is Necessary for Myogenic Differentiation. J. Agric Food Chem. 2021, 69, 3942–3951. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, A.; Ma, Y.; Zhang, X.; Pan, Q.; Luo, P.; Guo, H.; Wu, W.; Li, J.; Yu, T.; Liu, H. The Defects of Epigenetic Reprogramming in Dox-Dependent Porcine-iPSCs. Int. J. Mol. Sci. 2022, 23, 11941. https://doi.org/10.3390/ijms231911941
Jiang A, Ma Y, Zhang X, Pan Q, Luo P, Guo H, Wu W, Li J, Yu T, Liu H. The Defects of Epigenetic Reprogramming in Dox-Dependent Porcine-iPSCs. International Journal of Molecular Sciences. 2022; 23(19):11941. https://doi.org/10.3390/ijms231911941
Chicago/Turabian StyleJiang, Aiwen, Yangyang Ma, Xue Zhang, Qianqian Pan, Pengfei Luo, Hongyun Guo, Wangjun Wu, Juan Li, Tong Yu, and Honglin Liu. 2022. "The Defects of Epigenetic Reprogramming in Dox-Dependent Porcine-iPSCs" International Journal of Molecular Sciences 23, no. 19: 11941. https://doi.org/10.3390/ijms231911941
APA StyleJiang, A., Ma, Y., Zhang, X., Pan, Q., Luo, P., Guo, H., Wu, W., Li, J., Yu, T., & Liu, H. (2022). The Defects of Epigenetic Reprogramming in Dox-Dependent Porcine-iPSCs. International Journal of Molecular Sciences, 23(19), 11941. https://doi.org/10.3390/ijms231911941