
Contraction Band Necrosis with Dephosphorylated Connexin 43 in Rat Myocardium after Daily Cocaine Administration


Abstract
:1. Introduction
2. Results
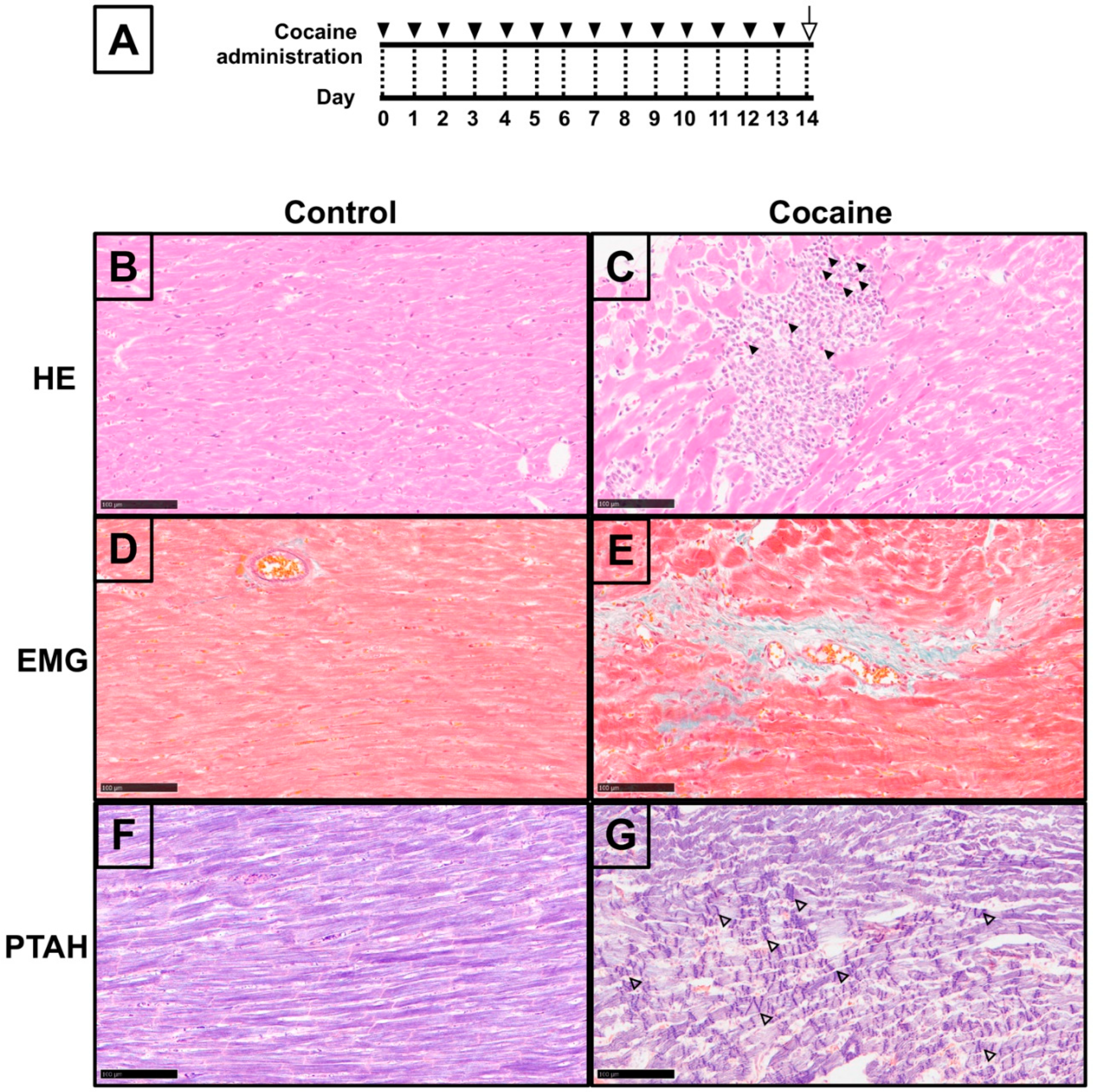
2.1. Pathological Changes in Myocardium after 14 Days of Cocaine Administration via Tail Vein
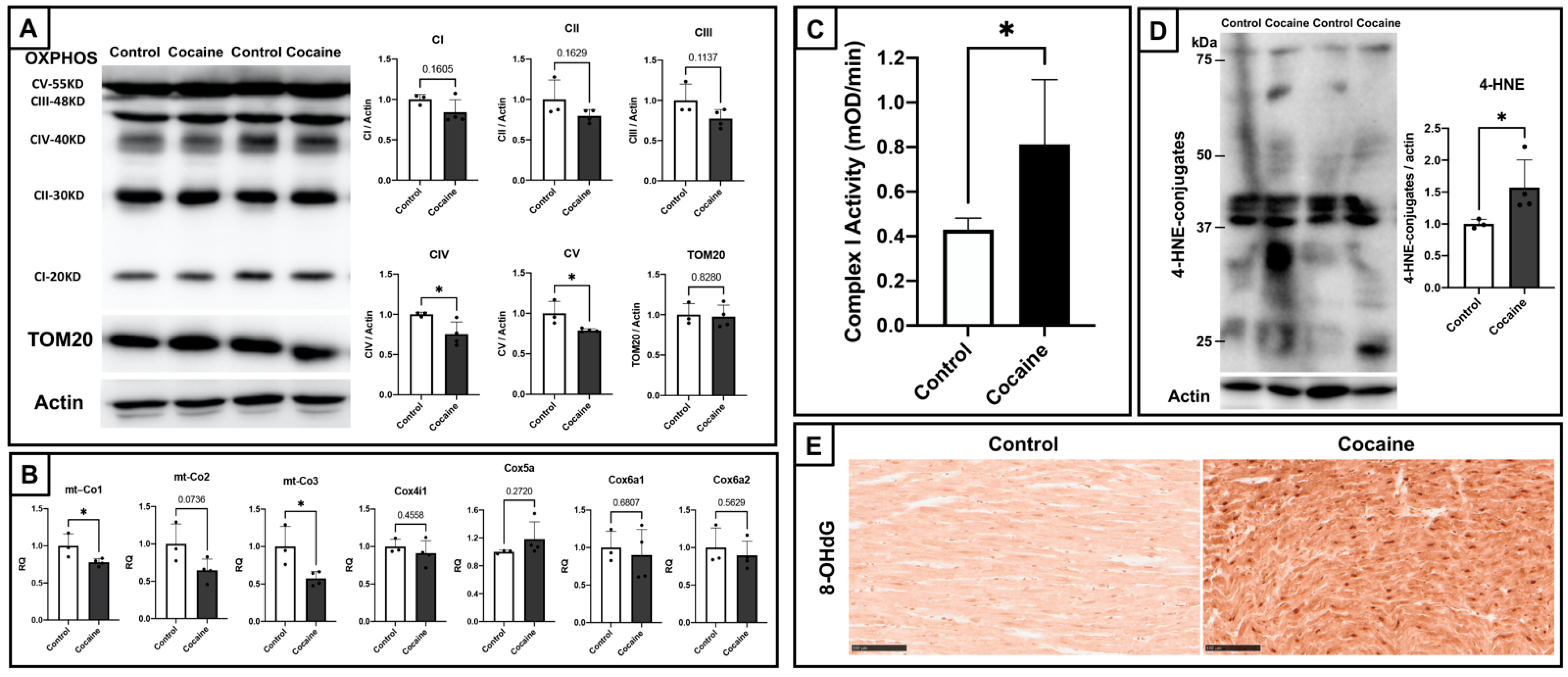
2.2. Cardiac Mitochondrial Dysfunction and Elevated Oxidative Stress in Myocardium
2.3. Electron Microscopic Analysis of Myocardium after 14 Days of Cocaine Administration via Tail Vein
2.4. Dephosphorylated Cardiac Cx43 and Its Distribution in Myocardium
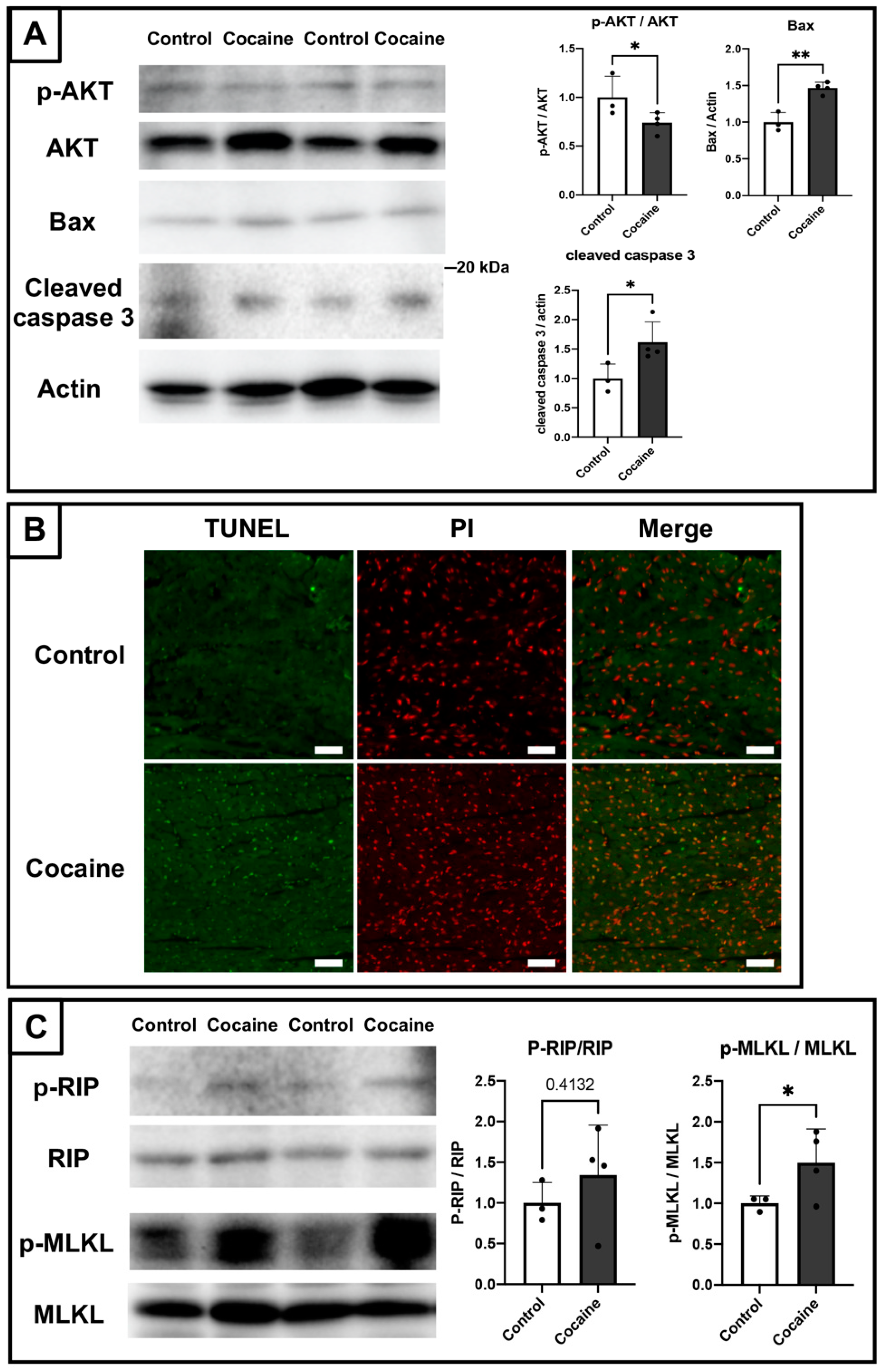
2.5. Involvement of Apoptosis and Necroptosis in the Damage to the Myocardium Caused by Cocaine Administration
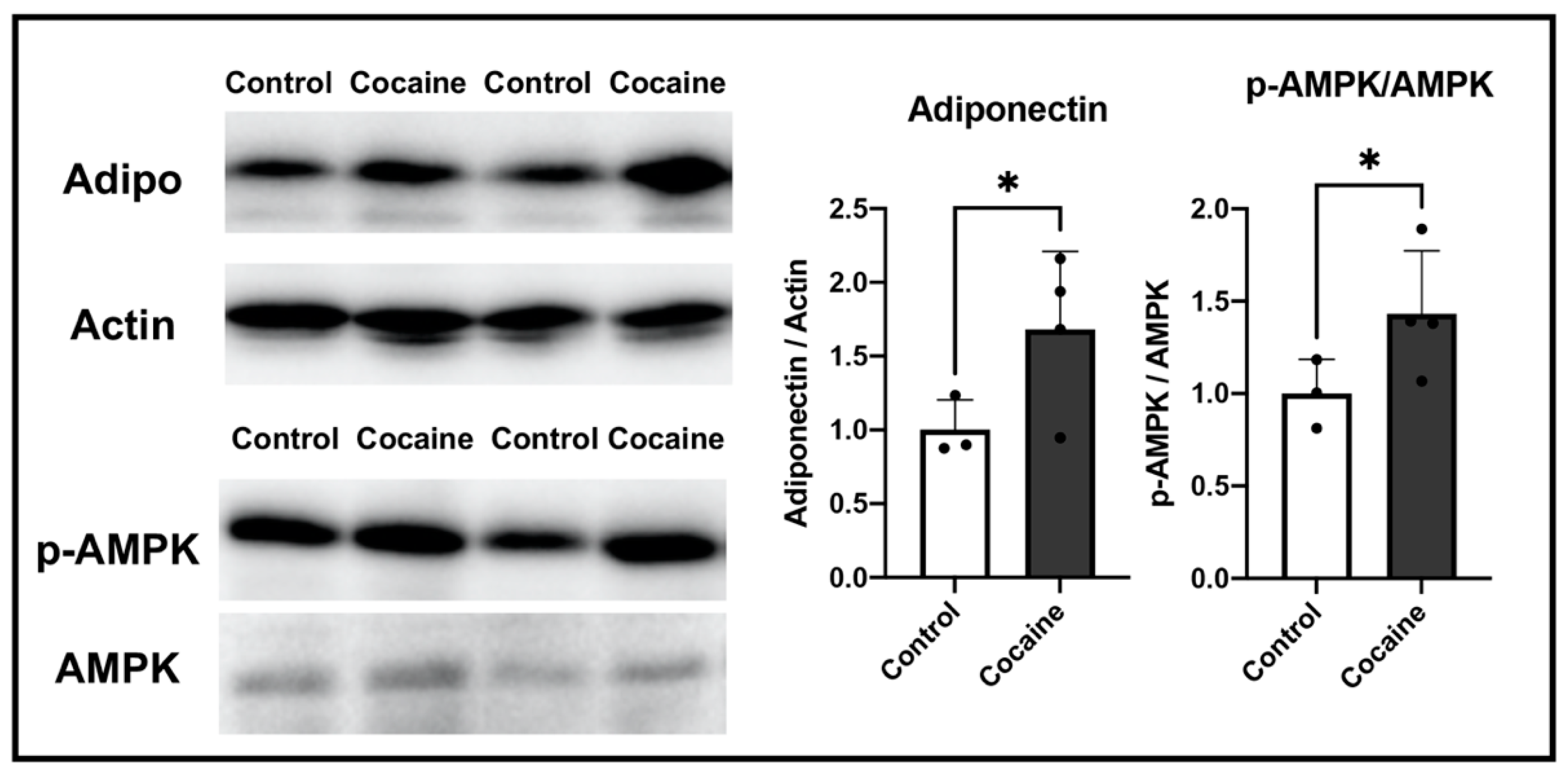
2.6. Possible Involvement of Adiponectin in Protecting the Myocardium after Cocaine Administration
3. Discussion
4. Materials and Methods
4.1. Animals and Cocaine Administration
4.2. Histological Analysis and Immunohistochemical Staining
4.3. Transmission Electron Microscopy
4.4. Mitochondrial Electron Transport Chain Activity Detection
4.5. Immunoblotting
4.6. Real-Time Reverse Transcriptase-Mediated PCR Analysis
4.7. TUNEL Analysis
4.8. Transcriptome Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Isner, J.M.; Estes, N.M., III; Thompson, P.D.; Costanzo-Nordin, M.R.; Subramanian, R.; Miller, G.; Katsas, G.; Sweeney, K.; Sturner, W.Q. Acute Cardiac Events Temporally Related to Cocaine Abuse. N. Engl. J. Med. 1986, 315, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Hale, S.; Alker, K.; Rezkalla, S. The Effects of Acute and Chronic Cocaine Use on the Heart. Circulation 1992, 85, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, K.; Luk, A.; Soor, G.S.; Abraham, J.R.; Leong, S.; Butany, J. Cocaine Cardiotoxicity: A Review of the Pathophysiology, Pathology, and Treatment Options. Am. J. Cardiovasc. Drugs 2009, 9, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Crumb, W.J., Jr.; Clarkson, C.W. Characterization of Cocaine-Induced Block of Cardiac Sodium Channels. Biophys. J. 1990, 57, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Pitts, W.R.; Lange, R.A.; Cigarroa, J.E.; Hillis, L.D. Cocaine-Induced Myocardial Ischemia and Infarction: Pathophysiology, Recognition, and Management. Prog. Cardiovasc. Dis. 1997, 40, 65–76. [Google Scholar] [CrossRef]
- Georgieva, E.; Karamalakova, Y.; Miteva, R.; Abrashev, H.; Nikolova, G. Oxidative Stress and Cocaine Intoxication as Start Points in the Pathology of Cocaine-Induced Cardiotoxicity. Toxics 2021, 9, 317. [Google Scholar] [CrossRef]
- Fineschi, V.; Wetli, C.V.; Paolo, M.D.; Baroldi, G. Myocardial Necrosis and Cocaine. A Quantitative Morphologic Study in 26 Cocaine-Associated Deaths. Int. J. Leg. Med. 1997, 110, 193–198. [Google Scholar] [CrossRef]
- Tazelaar, H.D.; Karch, S.B.; Stephens, B.G.; Billingham, M.E. Cocaine and the Heart. Hum. Pathol. 1987, 18, 195–199. [Google Scholar] [CrossRef]
- Karch, S.B.; Billingham, M.E. Myocardial Contraction Bands Revisited. Hum. Pathol. 1986, 17, 9–13. [Google Scholar] [CrossRef]
- Todd, G.L.; Baroldi, G.; Pieper, G.M.; Clayton, F.C.; Eliot, R.S. Experimental Catecholamine-Induced Myocardial Necrosis. I. Morphology, Quantification and Regional Distribution of Acute Contraction Band Lesions. J. Mol. Cell Cardiol. 1985, 17, 317–338. [Google Scholar] [CrossRef]
- Baroldi, G. Different Types of Myocardial Necrosis in Coronary Heart Disease: A Pathophysiologic Review of Their Functional Significance. Am. Heart J. 1975, 89, 742–752. [Google Scholar] [CrossRef]
- Karch, S.B. Resuscitation-Induced Myocardial Necrosis. Catecholamines and Defibrillation. Am. J. Forensic. Med. Pathol. 1987, 8, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Inserte, J.; Ruiz-Meana, M.; González, M.A.; Solares, J.; Juliá, M.; Barrabes, J.A.; Soler-Soler, J. Gap Junction Uncoupler Heptanol Prevents Cell-to-Cell Progression of Hypercontracture and Limits Necrosis During Myocardial Reperfusion. Circulation 1997, 96, 3579–3586. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Hofstaetter, B.; Piper, H.M.; Soler-Soler, J. Propagation of Cardiomyocyte Hypercontracture by Passage of Na(+) through Gap Junctions. Circ. Res. 1999, 85, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani-Ishida, K.; Unuma, K.; Yoshida, K. Ischemia Enhances Translocation of Connexin43 and Gap Junction Intercellular Communication, Thereby Propagating Contraction Band Necrosis after Reperfusion. Circ. J. 2009, 73, 1661–1668. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J.; Bruce, A.F.; Dupont, E.; Rothery, S. Remodelling of Gap Junctions and Connexin Expression in Diseased Myocardium. Cardiovasc. Res. 2008, 80, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R.; Görge, P.M.; Görbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 Is an Emerging Therapeutic Target in Ischemia/Reperfusion Injury, Cardioprotection and Neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of Connexin43 Phosphorylated at S325, S328 and S330 in Normoxic and Ischemic Heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef] [Green Version]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at S365 Is a Gatekeeper Event That Changes the Structure of Cx43 and Prevents Down-Regulation by Pkc. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary Trafficking Subunit Gja1-20k Protects Connexin-43 from Degradation and Limits Ventricular Arrhythmias. J. Clin. Invest. 2020, 130, 4858–4870. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, K.A.; Yamada, A.; Kleber, G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and Intracellular Redistribution of Ventricular Connexin43 During Electrical Uncoupling Induced by Ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yan, X.; Xue, J.; Zheng, Y.; Chen, M.; Sun, Z.; Liu, T.; Wang, C.; You, H.; Luo, D. Connexin43 Dephosphorylation at Serine 282 Is Associated with Connexin43-Mediated Cardiomyocyte Apoptosis. Cell Death Differ. 2019, 26, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.W. Role of Gap Junctions in Cardiac Conduction and Development: Insights from the Connexin Knockout Mice. Circ Res. 2000, 87, 346–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffitz, J.E. Arrhythmogenic Cardiomyopathy and Abnormalities of Cell-to-Cell Coupling. Heart Rhythm 2009, 6, S62–S655. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Adiponectin and Cardiovascular Health: An Update. Br. J. Pharmacol. 2012, 165, 574–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavuz, S.; Ece, A. Mean Platelet Volume as an Indicator of Disease Activity in Juvenile Sle. Clin. Rheumatol. 2014, 33, 637–641. [Google Scholar] [CrossRef]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar]
- Caselli, C.; D’Amico, A.; Cabiati, M.; Prescimone, T.; Del Ry, S.; Giannessi, D. Back to the heart: The protective role of adiponectin. Pharmacol. Res. 2014, 82, 9–20. [Google Scholar] [CrossRef]
- Leffler, K.E.; Abdel-Rahman, A.A. Restoration of Adiponectin-Connexin43 Signaling Mitigates Myocardial Inflammation and Dysfunction in Diabetic Female Rats. J. Cardiovasc. Pharmacol. 2020, 75, 259–267. [Google Scholar] [CrossRef]
- Liu, L.; Yan, M.; Yang, R.; Qin, X.; Chen, L.; Li, L.; Si, J.; Li, X.; Ma, K. Adiponectin Attenuates Lipopolysaccharide-Induced Apoptosis by Regulating the Cx43/Pi3k/Akt Pathway. Front. Pharmacol. 2021, 12, 644225. [Google Scholar] [CrossRef]
- Moritz, F.; Monteil, C.; Isabelle, M.; Bauer, F.; Renet, S.; Mulder, P.; Richard, V.; Thuillez, C. Role of Reactive Oxygen Species in Cocaine-Induced Cardiac Dysfunction. Cardiovasc. Res. 2003, 59, 834–843. [Google Scholar] [CrossRef]
- Fineschi, V.; Baroldi, G.; Centini, F.; Cerretani, D.; Fiaschi, A.I.; Micheli, L.; Parolini, M.; Turillazzi, E.; Giorgi, G. Markers of Cardiac Oxidative Stress and Altered Morphology after Intraperitoneal Cocaine Injection in a Rat Model. Int. J. Leg. Med. 2001, 114, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Todd, G.L.; Cullan, G.E.; Cullan, G.M. Isoproterenol-Induced Myocardial Necrosis and Membrane Permeability Alterations in the Isolated Perfused Rabbit Heart. Exp. Mol. Pathol. 1980, 33, 43–54. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome C Oxidase Dysfunction in Oxidative Stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Peng, S.K.; French, W.J.; Pelikan, P.C. Direct cocaine cardiotoxicity demonstrated by endomyocardial biopsy. Arch. Pathol. Lab. Med. 1989, 113, 842–845. [Google Scholar]
- Zhou, H.; Li, X.-M.; Meinkoth, J.; Pittman, R.N. Akt Regulates Cell Survival and Apoptosis at a Postmitochondrial Level. J. Cell Biol. 2000, 151, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Rickard, J.A.; Gerlic, M. The Diverse Role of Rip Kinases in Necroptosis and Inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef]
- Hirata, Y.; Kurobe, H.; Akaike, M.; Chikugo, F.; Hori, T.; Bando, Y.; Nishio, C.; Higashida, M.; Nakaya, Y.; Kitagawa, T.; et al. Enhanced Inflammation in Epicardial Fat in Patients with Coronary Artery Disease. Int. Heart J. 2011, 52, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, R.; Iglesias, M.J.; Gallego, R.; Raghay, K.; Eiras, S.; Rubio, J.; Diéguez, C.; Gualillo, O.; Gonzalez-Juanatey, J.R.; Lago, F. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett. 2005, 579, 5163–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, J.S.; Billingham, M.E.; Ginsburg, R.; Tsujimoto, G.; Lurie, K.G.; Hoffman, B.B. Cardiomyopathy in a Rat Model of Pheochromocytoma. Morphological and Functional Alterations. Am. J. Cardiovasc. Pathol. 1988, 1, 389–399. [Google Scholar] [PubMed]
- Virmani, R.; Robinowitz, M.; Smialek, J.E.; Smyth, D.F. Cardiovascular Effects of Cocaine: An Autopsy Study of 40 Patients. Am. Heart J. 1988, 115, 1068–1076. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid Turnover of Connexin43 in the Adult Rat Heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Görbe, A.; Varga, Z.V.; Kupai, K.; Bencsik, P.; Kocsis, G.F.; Csont, T.; Boengler, K.; Schulz, R.; Ferdinandy, P. Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: The role of connexin 43. Am. J. Physiol. Circ. Physiol. 2011, 300, H1907–H1913. [Google Scholar] [CrossRef] [Green Version]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.M.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Smith, K.; Yu, Q.; Miller, C.; Singh, K.; Sen, C.K. Mitochondrial connexin 43 in sex-dependent myocardial responses and estrogen-mediated cardiac protection following acute ischemia/reperfusion injury. Basic Res. Cardiol. 2019, 115, 1. [Google Scholar] [CrossRef]
- Shibata, R.; Sato, K.; Pimentel, D.R.; Takemura, Y.; Kihara, S.; Ohashi, K.; Funahashi, T.; Ouchi, N.; Walsh, K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2–dependent mechanisms. Nat. Med. 2005, 11, 1096–1103. [Google Scholar] [CrossRef] [Green Version]
- Essick, E.E.; Ouchi, N.; Wilson, R.M.; Ohashi, K.; Ghobrial, J.; Shibata, R.; Pimentel, D.R.; Sam, F. Adiponectin mediates cardioprotection in oxidative stress-induced cardiac myocyte remodeling. Am. J. Physiol. Circ. Physiol. 2011, 301, H984–H993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, R.; Ouchi, N.; Ito, M.; Kihara, S.; Shiojima, I.; Pimentel, D.R.; Kumada, M.; Sato, K.; Schiekofer, S.; Ohashi, K.; et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 2004, 10, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- You, Z.B.; Wang, B.; Gardner, E.L.; Wise, R.A. Cocaine and Cocaine Expectancy Increase Growth Hormone, Ghrelin, Glp-1, Igf-1, Adiponectin, and Corticosterone While Decreasing Leptin, Insulin, Gip, and Prolactin. Pharmacol. Biochem. Behav. 2019, 176, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Unuma, K.; Funakoshi, T.; Aki, T.; Uemura, K. Altered cardiac mitochondrial dynamics and biogenesis in rat after short-term cocaine administration. Sci. Rep. 2021, 11, 24129. [Google Scholar] [CrossRef]
- Aso, Y.; Yamamoto, R.; Wakabayashi, S.; Uchida, T.; Takayanagi, K.; Takebayashi, K.; Okuno, T.; Inoue, T.; Node, K.; Tobe, T.; et al. Comparison of Serum High-Molecular Weight (Hmw) Adiponectin with Total Adiponectin Concentrations in Type 2 Diabetic Patients with Coronary Artery Disease Using a Novel Enzyme-Linked Immunosorbent Assay to Detect Hmw Adiponectin. Diabetes 2006, 55, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Heard, K.; Palmer, R.; Zahniser, N.R. Mechanisms of Acute Cocaine Toxicity. Open Pharmacol. J. 2008, 2, 70–78. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, T.; Furukawa, M.; Aki, T.; Uemura, K. Repeated exposure of cocaine alters mitochondrial dynamics in mouse neuroblastoma Neuro2a. NeuroToxicology 2019, 75, 70–77. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Change | Description | |
|---|---|---|---|
| 1 | Adipoq | 14.28 | adiponectin |
| 2 | Ttn | 9.55 | titin |
| 3 | Car3 | 6.1 | Carbonic anhydrase 3 |
| 4 | Tnnt2 | 5.51 | Troponin T type 2 |
| 5 | Mb | 5.39 | myoglobin |
| 6 | Alox15 | 4.92 | Arachidonate 15-lipoxygenase |
| 7 | Abcb1b | 4.57 | ATP-binding cassette, subfamily B, member 1B |
| 8 | Myh6 | 4.19 | Myosin heavy chain 6 |
| 9 | Myl7 | 3.83 | Myosin light chain 7 |
| 10 | Lcn2 | 3.63 | lipocalin2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, S.; Unuma, K.; Funakoshi, T.; Aki, T.; Uemura, K. Contraction Band Necrosis with Dephosphorylated Connexin 43 in Rat Myocardium after Daily Cocaine Administration. Int. J. Mol. Sci. 2022, 23, 11978. https://doi.org/10.3390/ijms231911978
Wen S, Unuma K, Funakoshi T, Aki T, Uemura K. Contraction Band Necrosis with Dephosphorylated Connexin 43 in Rat Myocardium after Daily Cocaine Administration. International Journal of Molecular Sciences. 2022; 23(19):11978. https://doi.org/10.3390/ijms231911978
Chicago/Turabian StyleWen, Shuheng, Kana Unuma, Takeshi Funakoshi, Toshihiko Aki, and Koichi Uemura. 2022. "Contraction Band Necrosis with Dephosphorylated Connexin 43 in Rat Myocardium after Daily Cocaine Administration" International Journal of Molecular Sciences 23, no. 19: 11978. https://doi.org/10.3390/ijms231911978
APA StyleWen, S., Unuma, K., Funakoshi, T., Aki, T., & Uemura, K. (2022). Contraction Band Necrosis with Dephosphorylated Connexin 43 in Rat Myocardium after Daily Cocaine Administration. International Journal of Molecular Sciences, 23(19), 11978. https://doi.org/10.3390/ijms231911978