
Diverse Roles for a Conserved DNA-Methyltransferase in the Entomopathogenic Bacterium Xenorhabdus

Abstract
:1. Introduction
2. Results
2.1. MTases Repertoire in the Genus Xenorhabdus
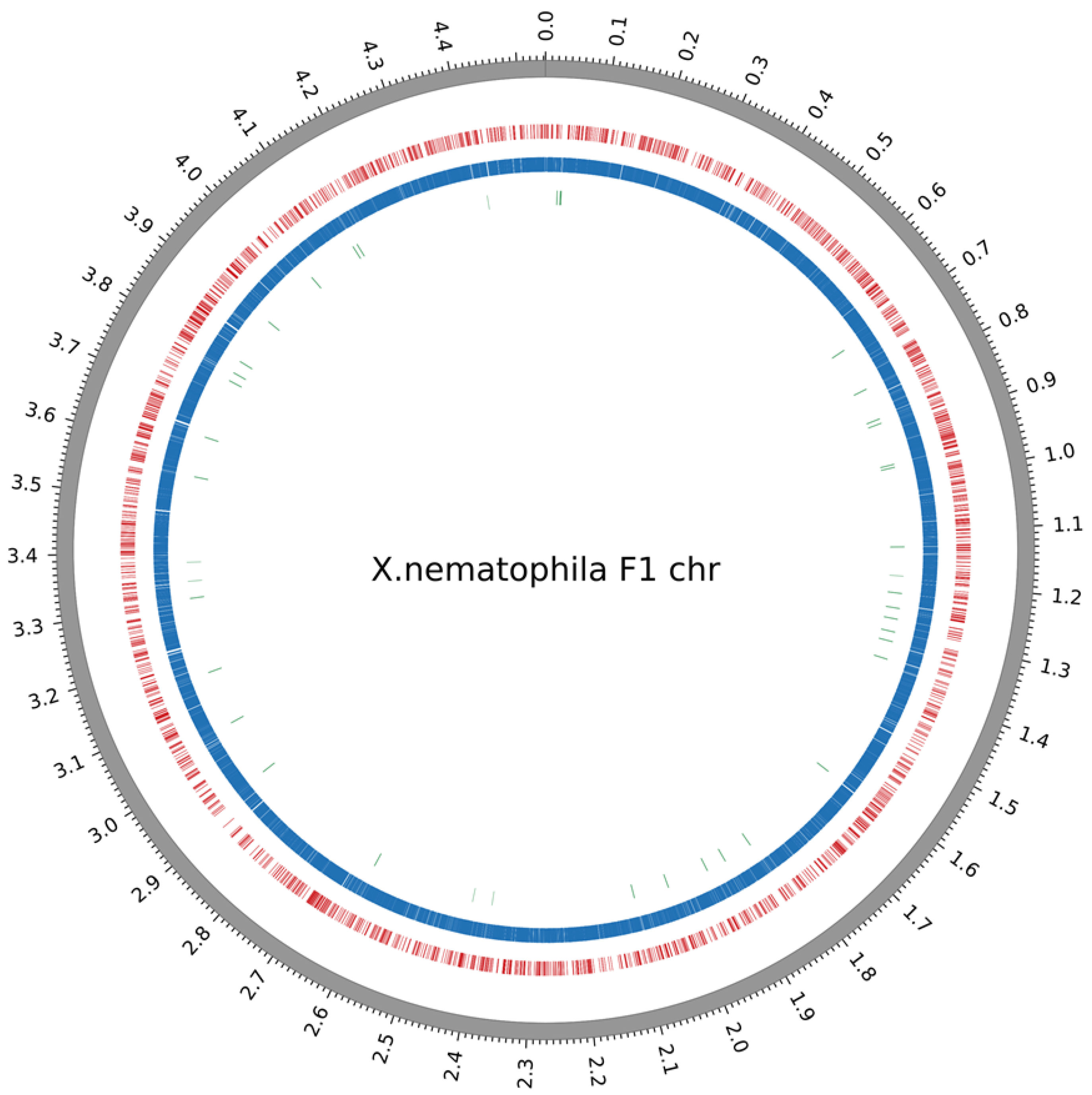
2.2. Methylome Analysis in Xenorhabdus
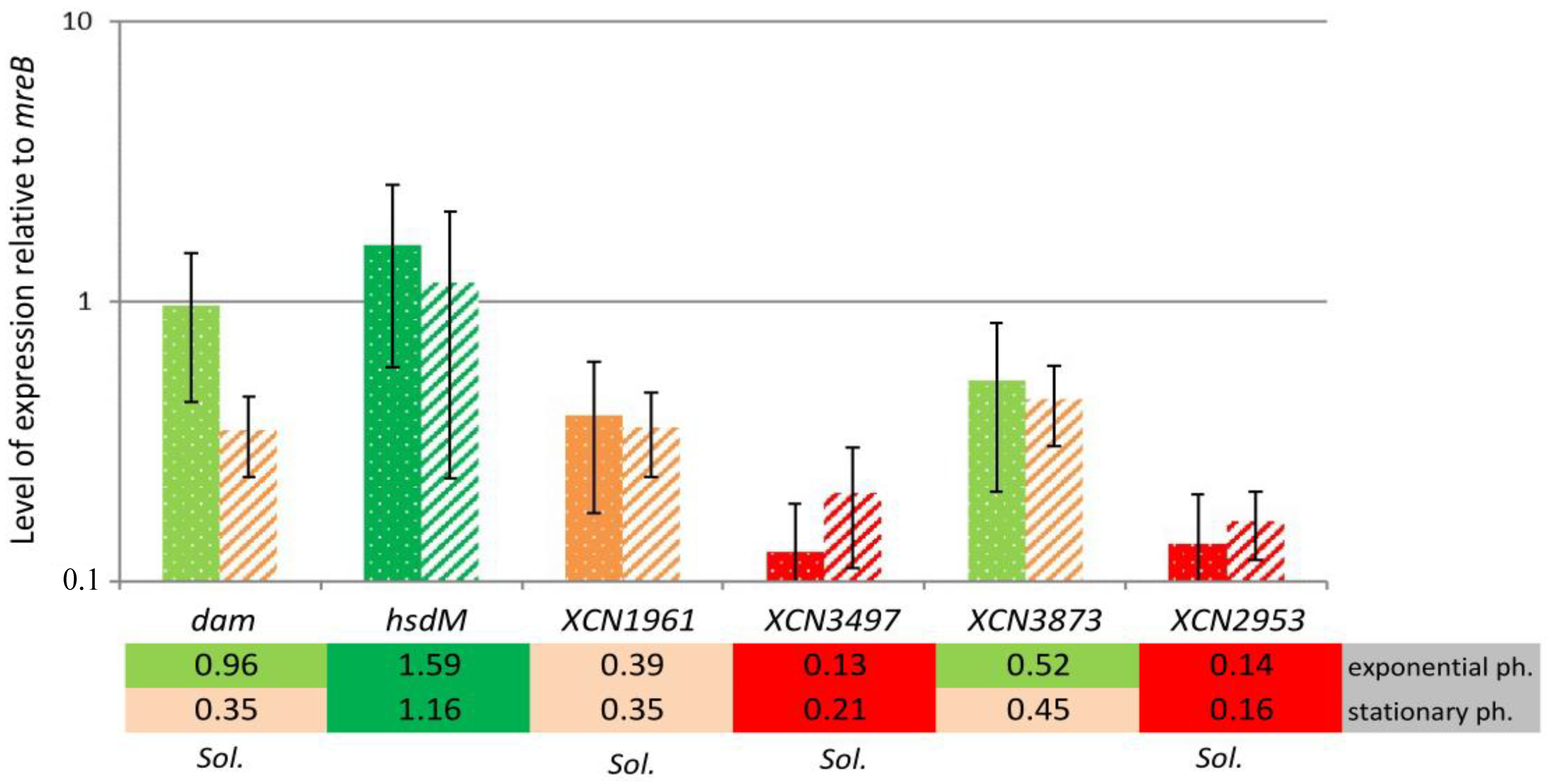
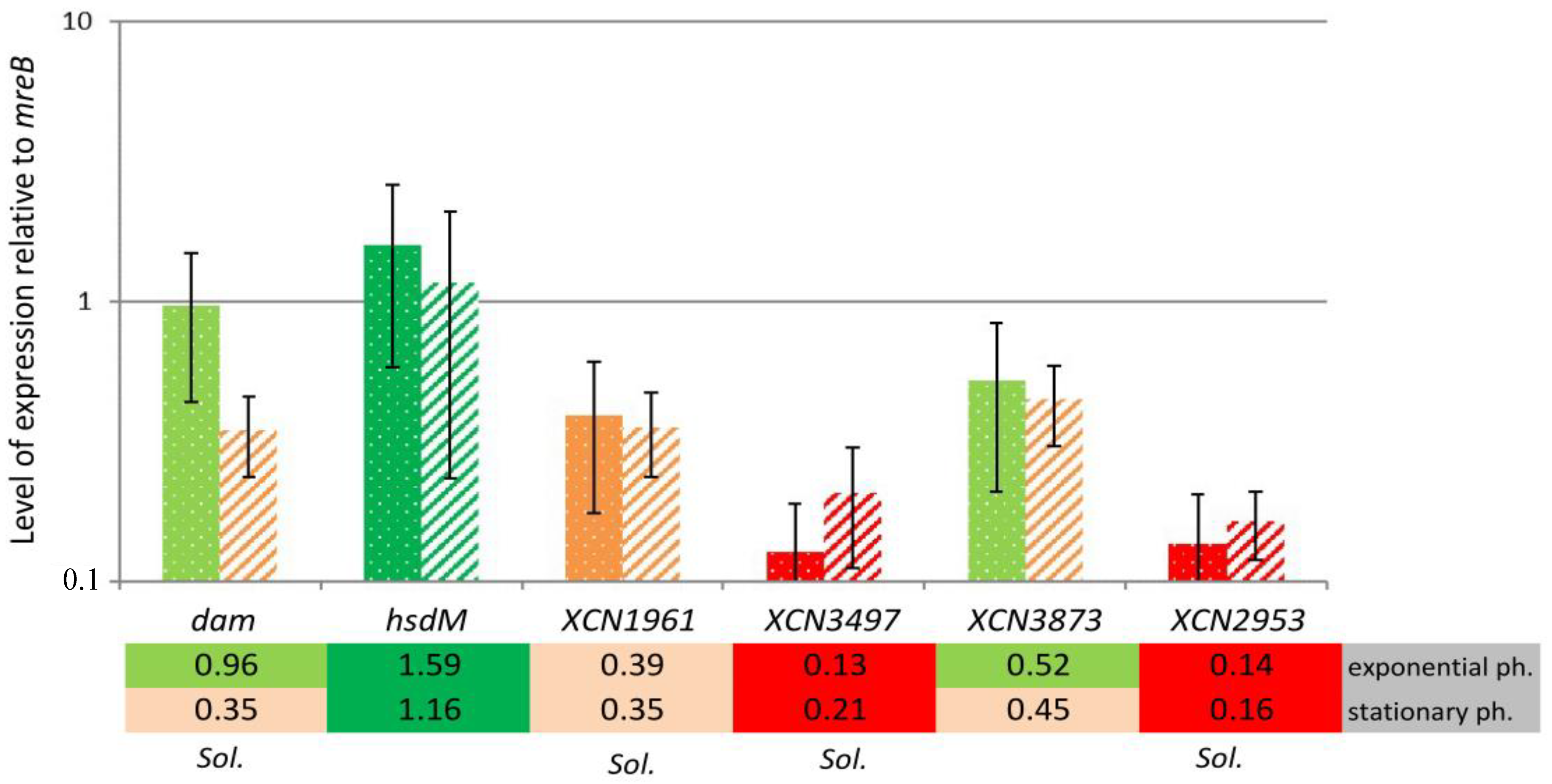
2.3. MTase Expression
2.4. Unmethylated GATC Sites Are More Frequently Associated with Intergenic Regions
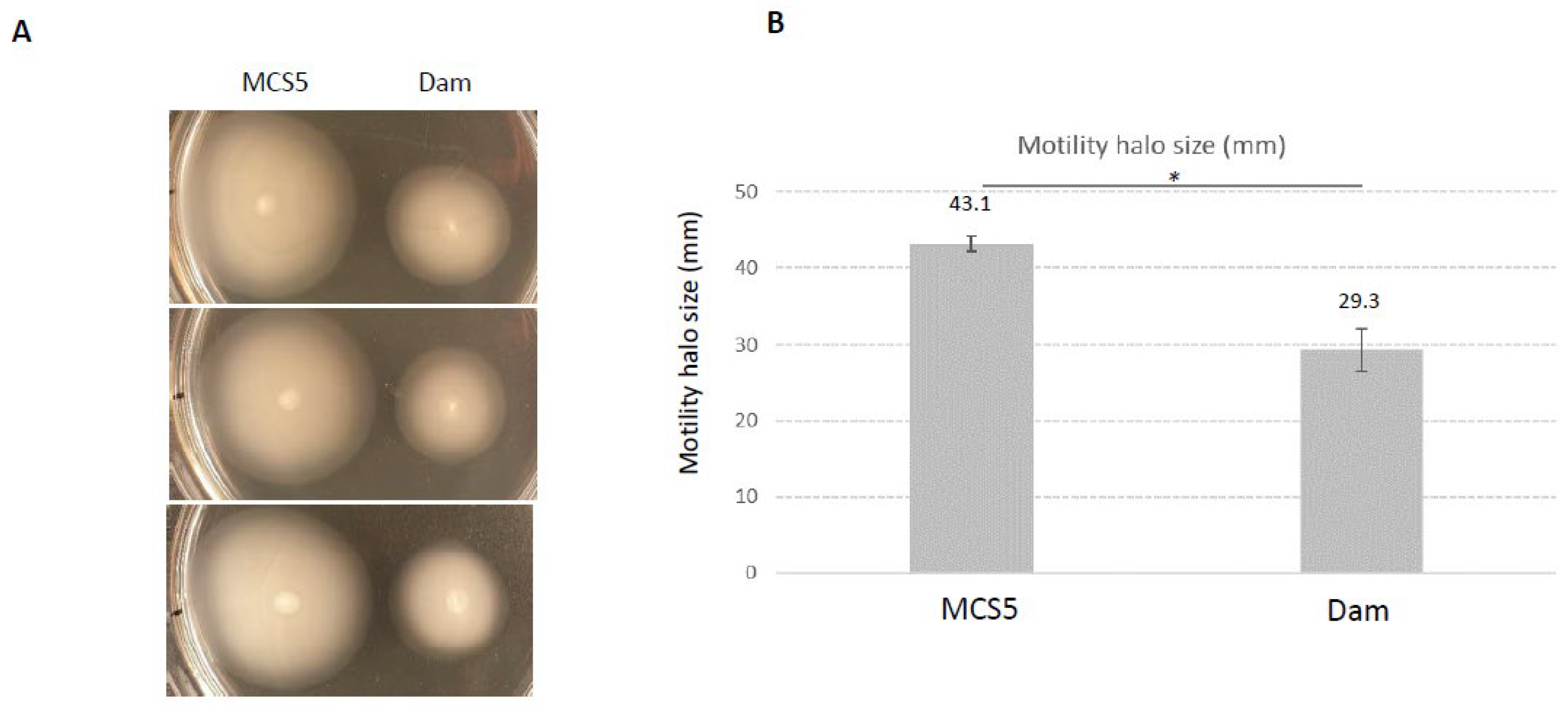
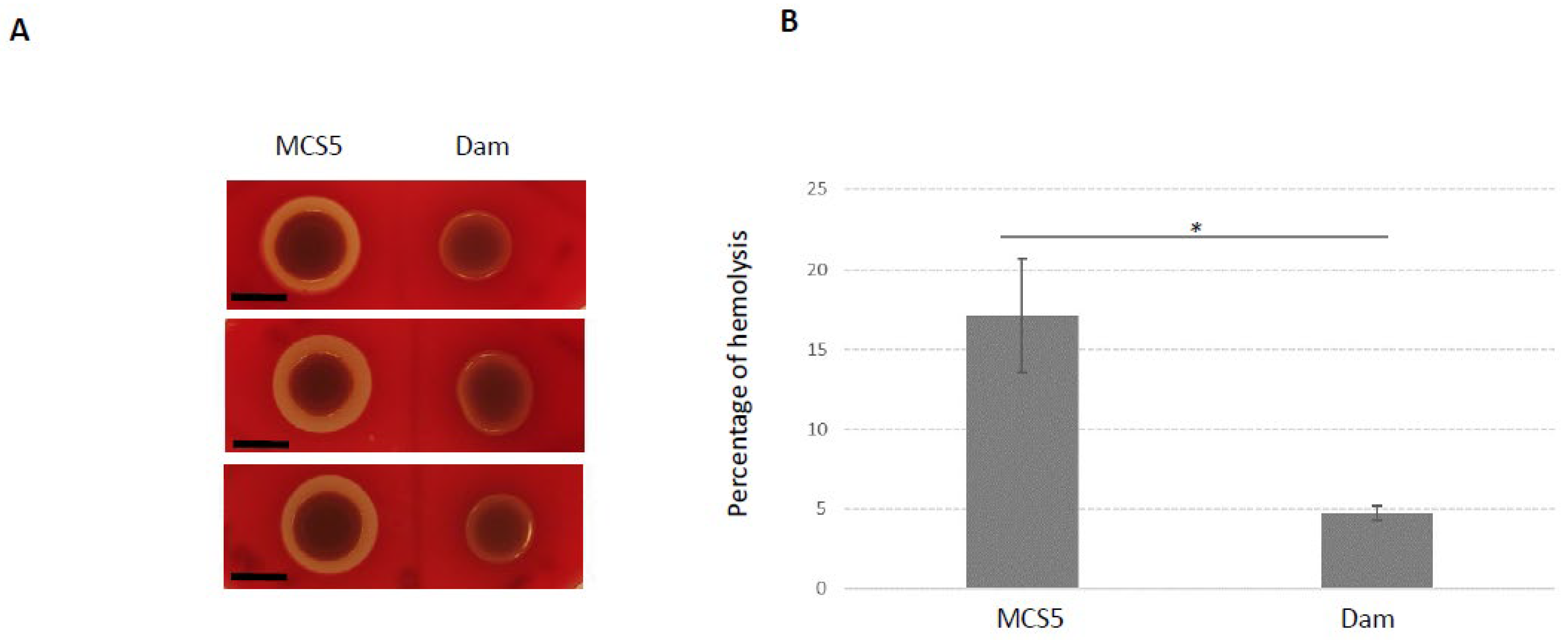
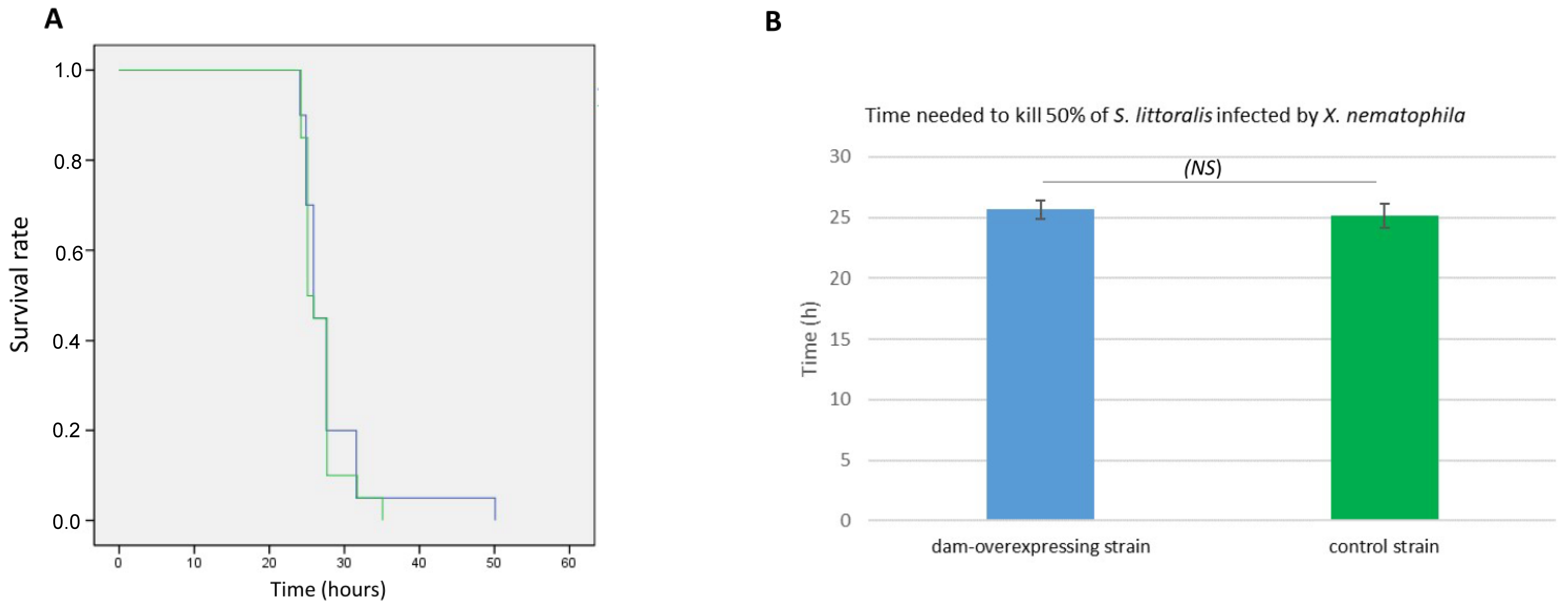
2.5. Phenotypes Associated with Dam Overexpression
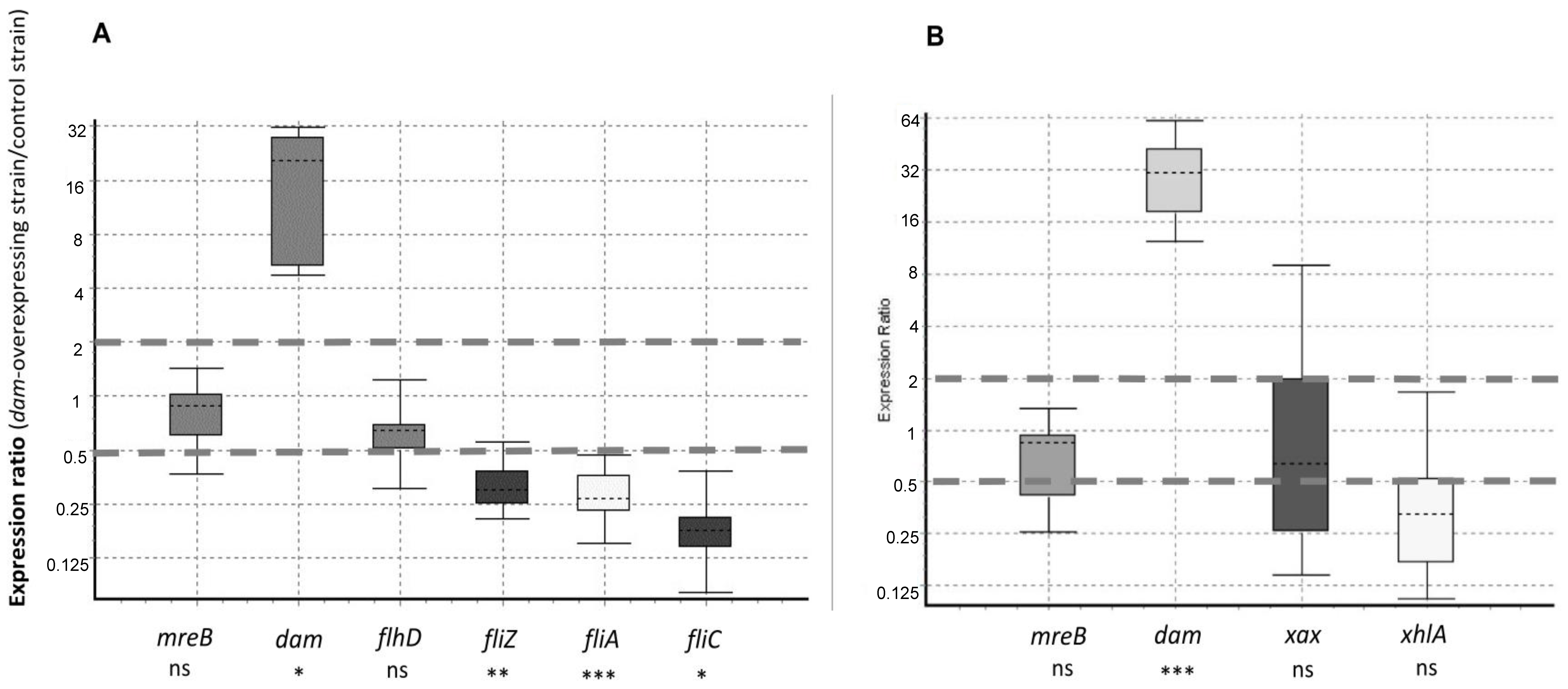
2.6. Flagellar Genes Are Downregulated in the X. nematophila dam-Overexpressing Strain
3. Discussion
4. Materials and Methods
4.1. Strains and Growth Conditions
4.2. Distribution of DNA-Methyltransferases in Xenorhabdus by In Silico Analysis
4.3. Genome Sequencing and DNA Methylation Detection and Motifs Identification
4.4. Nucleic Acid Manipulations
4.5. Methylation-Sensitive Restriction Enzyme (MSRE) PCR Analysis
4.6. Phenotype Analysis of X. nematophila Overexpressing Dam
4.7. Insect Virulence Assay
4.8. RNA Preparation and RT-qPCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Sanchez-Romero, M.A.; Casadesus, J. The bacterial epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Billen, D. Methylation of the bacterial chromosome: An event at the “replication point”? J. Mol. Biol. 1968, 31, 477–486. [Google Scholar] [CrossRef]
- Anton, B.P.; Roberts, R.J. Beyond Restriction Modification: Epigenomic Roles of DNA Methylation in Prokaryotes. Annu. Rev. Microbiol. 2021, 75, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinus, M.G.; Lobner-Olesen, A. DNA Methylation. EcoSal Plus 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE--a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef]
- Lobner-Olesen, A.; Skovgaard, O.; Marinus, M.G. Dam methylation: Coordinating cellular processes. Curr. Opin. Microbiol. 2005, 8, 154–160. [Google Scholar] [CrossRef]
- Casadesus, J. Bacterial DNA Methylation and Methylomes. Adv. Exp. Med. Biol. 2016, 945, 35–61. [Google Scholar] [CrossRef]
- Casadesus, J.; Low, D. Epigenetic gene regulation in the bacterial world. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [Green Version]
- Casadesus, J.; Low, D.A. Programmed heterogeneity: Epigenetic mechanisms in bacteria. J. Biol. Chem. 2013, 288, 13929–13935. [Google Scholar] [CrossRef] [Green Version]
- Low, D.A.; Casadesus, J. Clocks and switches: Bacterial gene regulation by DNA adenine methylation. Curr. Opin. Microbiol. 2008, 11, 106–112. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [Green Version]
- Weigel, W.A.; Dersch, P. Phenotypic heterogeneity: A bacterial virulence strategy. Microbes Infect. 2018, 20, 570–577. [Google Scholar] [CrossRef]
- Garcia-Del Portillo, F.; Pucciarelli, M.G.; Casadesus, J. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 11578–11583. [Google Scholar] [CrossRef] [Green Version]
- Heithoff, D.M.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. An essential role for DNA adenine methylation in bacterial virulence. Science 1999, 284, 967–970. [Google Scholar] [CrossRef]
- Wu, H.; Lippmann, J.E.; Oza, J.P.; Zeng, M.; Fives-Taylor, P.; Reich, N.O. Inactivation of DNA adenine methyltransferase alters virulence factors in Actinobacillus actinomycetemcomitans. Oral Microbiol. Immunol. 2006, 21, 238–244. [Google Scholar] [CrossRef]
- Erova, T.E.; Fadl, A.A.; Sha, J.; Khajanchi, B.K.; Pillai, L.L.; Kozlova, E.V.; Chopra, A.K. Mutations within the catalytic motif of DNA adenine methyltransferase (Dam) of Aeromonas hydrophila cause the virulence of the Dam-overproducing strain to revert to that of the wild-type phenotype. Infect. Immun. 2006, 74, 5763–5772. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.E.; Jarisch, J.; Smith, A.L. Inactivation of deoxyadenosine methyltransferase (dam) attenuates Haemophilus influenzae virulence. Mol. Microbiol. 2004, 53, 651–664. [Google Scholar] [CrossRef]
- Mehling, J.S.; Lavender, H.; Clegg, S. A Dam methylation mutant of Klebsiella pneumoniae is partially attenuated. FEMS Microbiol. Lett. 2007, 268, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Paulsen, D.B.; Scruggs, D.W.; Banes, M.M.; Reeks, B.Y.; Lawrence, M.L. Alteration of DNA adenine methylase (Dam) activity in Pasteurella multocida causes increased spontaneous mutation frequency and attenuation in mice. Microbiology 2003, 149, 2283–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julio, S.M.; Heithoff, D.M.; Provenzano, D.; Klose, K.E.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 2001, 69, 7610–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, V.L.; Oyston, P.C.; Titball, R.W. A dam mutant of Yersinia pestis is attenuated and induces protection against plague. FEMS Microbiol. Lett. 2005, 252, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Demarre, G.; Chattoraj, D.K. DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet. 2010, 6, e1000939. [Google Scholar] [CrossRef] [Green Version]
- Payelleville, A.; Lanois, A.; Gislard, M.; Dubois, E.; Roche, D.; Cruveiller, S.; Givaudan, A.; Brillard, J. DNA Adenine Methyltransferase (Dam) Overexpression Impairs Photorhabdus luminescens Motility and Virulence. Front. Microbiol. 2017, 8, 1671. [Google Scholar] [CrossRef]
- Atack, J.M.; Tan, A.; Bakaletz, L.O.; Jennings, M.P.; Seib, K.L. Phasevarions of Bacterial Pathogens: Methylomics Sheds New Light on Old Enemies. Trends Microbiol. 2018, 26, 715–726. [Google Scholar] [CrossRef]
- Heusipp, G.; Falker, S.; Schmidt, M.A. DNA adenine methylation and bacterial pathogenesis. Int. J. Med. Microbiol. 2007, 297, 1–7. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Boemare, N.E. A numerical taxonomic study of the genus Xenorhabdus (Enterobacteriaceae) and proposed elevation of the subspecies of X. nematophilus to species. J. Gen. Microbiol. 1988, 134, 1835–1845. [Google Scholar] [CrossRef]
- Payelleville, A.; Legrand, L.; Ogier, J.C.; Roques, C.; Roulet, A.; Bouchez, O.; Mouammine, A.; Givaudan, A.; Brillard, J. The complete methylome of an entomopathogenic bacterium reveals the existence of loci with unmethylated Adenines. Sci. Rep. 2018, 8, 12091. [Google Scholar] [CrossRef]
- Payelleville, A.; Brillard, J. Novel Identification of Bacterial Epigenetic Regulations Would Benefit From a Better Exploitation of Methylomic Data. Front. Microbiol. 2021, 12, 685670. [Google Scholar] [CrossRef]
- Lanois, A.; Pages, S.; Bourot, S.; Canoy, A.S.; Givaudan, A.; Gaudriault, S. Transcriptional analysis of a Photorhabdus sp. variant reveals transcriptional control of phenotypic variation and multifactorial pathogenicity in insects. Appl. Environ. Microbiol. 2011, 77, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Brillard, J.; Duchaud, E.; Boemare, N.; Kunst, F.; Givaudan, A. The PhlA hemolysin from the entomopathogenic bacterium Photorhabdus luminescens belongs to the two-partner secretion family of hemolysins. J. Bacteriol. 2002, 184, 3871–3878. [Google Scholar] [CrossRef] [Green Version]
- Brillard, J.; Ribeiro, C.; Boemare, N.; Brehelin, M.; Givaudan, A. Two distinct hemolytic activities in Xenorhabdus nematophila are active against immunocompetent insect cells. Appl. Environ. Microbiol. 2001, 67, 2515–2525. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Colson, C.; Van Pel, A. DNA restriction and modification systems in Salmonella. I. SA and SB, two Salmonella typhimurium systems determined by genes with a chromosomal location comparable to that of the Escherichia coli hsd genes. Mol. Gen. Genet. 1974, 129, 325–337. [Google Scholar] [CrossRef]
- Blyn, L.B.; Braaten, B.A.; Low, D.A. Regulation of pap pilin phase variation by a mechanism involving differential dam methylation states. EMBO J. 1990, 9, 4045–4054. [Google Scholar] [CrossRef]
- Casadesus, J.; Maldonado, R. Genomic imprinting in microorganisms. Microbiologia 1990, 6, 1–10. [Google Scholar]
- Erill, I.; Puigvert, M.; Legrand, L.; Guarischi-Sousa, R.; Vandecasteele, C.; Setubal, J.C.; Genin, S.; Guidot, A.; Valls, M. Comparative Analysis of Ralstonia solanacearum Methylomes. Front. Plant Sci. 2017, 8, 504. [Google Scholar] [CrossRef] [Green Version]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Ribis, J.W.; Garrett, E.M.; Trzilova, D.; Kim, A.; Sekulovic, O.; Mead, E.A.; Pak, T.; Zhu, S.; Deikus, G.; et al. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol. 2020, 5, 166–180. [Google Scholar] [CrossRef]
- Horton, J.R.; Liebert, K.; Bekes, M.; Jeltsch, A.; Cheng, X. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J. Mol. Biol. 2006, 358, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Cohen, N.R.; Ross, C.A.; Jain, S.; Shapiro, R.S.; Gutierrez, A.; Belenky, P.; Li, H.; Collins, J.J. A role for the bacterial GATC methylome in antibiotic stress survival. Nat. Genet. 2016, 48, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Cota, I.; Bunk, B.; Sproer, C.; Overmann, J.; Konig, C.; Casadesus, J. OxyR-dependent formation of DNA methylation patterns in OpvABOFF and OpvABON cell lineages of Salmonella enterica. Nucleic Acids Res. 2016, 44, 3595–3609. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, J.S.; Anderson, C.E.; Wang, L.; Modliszewski, J.L.; Chen, W.; Schott, B.H.; Devos, N.; Ko, D.C. Integration of the Salmonella Typhimurium Methylome and Transcriptome Reveals That DNA Methylation and Transcriptional Regulation Are Largely Decoupled under Virulence-Related Conditions. mBio 2022, 13, e0346421. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Olivenza, D.R.; Gutiérrez, G.; Casadesús, J. Contribution of DNA adenine methylation to gene expression heterogeneity in Salmonella enterica. Nucleic Acids Res. 2020, 48, 11857–11867. [Google Scholar] [CrossRef]
- Braaten, B.A.; Nou, X.; Kaltenbach, L.S.; Low, D.A. Methylation patterns in pap regulatory DNA control pyelonephritis-associated pili phase variation in E. coli. Cell 1994, 76, 577–588. [Google Scholar] [CrossRef]
- Braaten, B.A.; Platko, J.V.; van der Woude, M.W.; Simons, B.H.; de Graaf, F.K.; Calvo, J.M.; Low, D.A. Leucine-responsive regulatory protein controls the expression of both the pap and fan pili operons in Escherichia coli. Proc. Natl. Acad. Sci. USA 1992, 89, 4250–4254. [Google Scholar] [CrossRef] [Green Version]
- Brunet, Y.R.; Bernard, C.S.; Gavioli, M.; Lloubes, R.; Cascales, E. An epigenetic switch involving overlapping fur and DNA methylation optimizes expression of a type VI secretion gene cluster. PLoS Genet. 2011, 7, e1002205. [Google Scholar] [CrossRef] [Green Version]
- Henderson, I.R.; Owen, P. The major phase-variable outer membrane protein of Escherichia coli structurally resembles the immunoglobulin A1 protease class of exported protein and is regulated by a novel mechanism involving Dam and OxyR. J. Bacteriol. 1999, 181, 2132–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadbent, S.E.; Davies, M.R.; van der Woude, M.W. Phase variation controls expression of Salmonella lipopolysaccharide modification genes by a DNA methylation-dependent mechanism. Mol. Microbiol. 2010, 77, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Cowles, K.N.; Cowles, C.E.; Richards, G.R.; Martens, E.C.; Goodrich-Blair, H. The global regulator Lrp contributes to mutualism, pathogenesis and phenotypic variation in the bacterium Xenorhabdus nematophila. Cell Microbiol. 2007, 9, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Lanois, A.; Jubelin, G.; Givaudan, A. FliZ, a flagellar regulator, is at the crossroads between motility, haemolysin expression and virulence in the insect pathogenic bacterium Xenorhabdus. Mol. Microbiol. 2008, 68, 516–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Li, J.; Barnes, I.H.; Baltzegar, D.A.; Pajaniappan, M.; Cullen, T.W.; Trent, M.S.; Burns, C.M.; Thompson, S.A. Role of the Campylobacter jejuni Cj1461 DNA methyltransferase in regulating virulence characteristics. J. Bacteriol. 2008, 190, 6524–6529. [Google Scholar] [CrossRef] [Green Version]
- Shell, S.S.; Prestwich, E.G.; Baek, S.H.; Shah, R.R.; Sassetti, C.M.; Dedon, P.C.; Fortune, S.M. DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003419. [Google Scholar] [CrossRef] [Green Version]
- Heithoff, D.M.; Enioutina, E.Y.; Daynes, R.A.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. Salmonella DNA adenine methylase mutants confer cross-protective immunity. Infect. Immun. 2001, 69, 6725–6730. [Google Scholar] [CrossRef] [Green Version]
- Julio, S.M.; Heithoff, D.M.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA adenine methylase overproduction in Yersinia pseudotuberculosis alters YopE expression and secretion and host immune responses to infection. Infect. Immun. 2002, 70, 1006–1009. [Google Scholar] [CrossRef] [Green Version]
- Givaudan, A.; Lanois, A. FlhDC, the flagellar master operon of Xenorhabdus nematophilus: Requirement for motility, lipolysis, extracellular hemolysis, and full virulence in insects. J. Bacteriol. 2000, 182, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Givaudan, A.; Lanois, A. Flagellar Regulation and Virulence in the Entomopathogenic Bacteria-Xenorhabdus nematophila and Photorhabdus luminescens. Curr. Top. Microbiol. Immunol. 2016, 402, 39–51. [Google Scholar] [CrossRef]
- Lanois, A.; Ogier, J.C.; Gouzy, J.; Laroui, C.; Rouy, Z.; Givaudan, A.; Gaudriault, S. Draft Genome Sequence and Annotation of the Entomopathogenic Bacterium Xenorhabdus nematophila Strain F1. Genome Announc 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Paulick, A.; Koerdt, A.; Lassak, J.; Huntley, S.; Wilms, I.; Narberhaus, F.; Thormann, K.M. Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol. Microbiol. 2009, 71, 836–850. [Google Scholar] [CrossRef]
- Lobner-Olesen, A.; von Freiesleben, U. Chromosomal replication incompatibility in Dam methyltransferase deficient Escherichia coli cells. EMBO J. 1996, 15, 5999–6008. [Google Scholar] [CrossRef]
- Kovach, M.E.; Elzer, P.H.; Hill, D.S.; Robertson, G.T.; Farris, M.A.; Roop, R.M., 2nd; Peterson, K.M. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 1995, 166, 175–176. [Google Scholar] [CrossRef]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Brillard, J.; Lereclus, D. Characterization of a small PlcR-regulated gene co-expressed with cereolysin O. BMC Microbiol. 2007, 7, 52. [Google Scholar] [CrossRef]
- Payelleville, A.; Blackburn, D.; Lanois, A.; Pages, S.; Cambon, M.C.; Ginibre, N.; Clarke, D.J.; Givaudan, A.; Brillard, J. Role of the Photorhabdus Dam methyltransferase during interactions with its invertebrate hosts. PLoS ONE 2019, 14, e0212655. [Google Scholar] [CrossRef]
- Boemare, N.E.; Akhurst, R.J. Biochemical and physiological characterization of colony form variants in Xenorhabdus spp. (Enterobacteriaceae). J Gen. Microbiol. 1988, 134, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Brillard, J.; Boyer-Giglio, M.H.; Boemare, N.; Givaudan, A. Holin locus characterisation from lysogenic Xenorhabdus nematophila and its involvement in Escherichia coli SheA haemolytic phenotype. FEMS Microbiol. Lett. 2003, 218, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Jubelin, G.; Lanois, A.; Severac, D.; Rialle, S.; Longin, C.; Gaudriault, S.; Givaudan, A. FliZ is a global regulatory protein affecting the expression of flagellar and virulence genes in individual Xenorhabdus nematophila bacterial cells. PLoS Genet. 2013, 9, e1003915. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motif 1 | Fraction | nDetected | nGenome | Mean Score | Mean Ipd Ratio | Mean Coverage | Objective Score |
|---|---|---|---|---|---|---|---|
| Motifs in X. nematophila F1 | |||||||
| GATC | 0.998 | 31,731 | 31,808 | 95.6 | 5.52 | 56.9 | 3,028,101 |
| CAGNNNNNGTG/ | 1.000 | 2021 | 2022 | 90.9 | 6.36 | 56.4 | 183,562 |
| CACNNNNNCTG | 1.000 | 2021 | 2022 | 87.5 | 5.32 | 56.7 | 176,691 |
| AANNNCCGGGNNNNNGA 2 | 0.760 | 73 | 96 | 49.7 | 3.49 | 49.7 | 2838 |
| Motifs in X. kozodoii FR48 | |||||||
| GATC | 0.996 | 33,246 | 33,380 | 124.2 | 7.41 | 77.5 | 4,114,426 |
| TTCANNNNNNGTG/ | 1.000 | 675 | 675 | 105.3 | 6.98 | 76.7 | 71,110 |
| CACNNNNNNTGAA 2 | 1.000 | 675 | 675 | 92.7 | 5.74 | 76.0 | 61,087 |
| CATCNNNNNNCTC/ | 0.991 | 425 | 429 | 111.2 | 7.08 | 74.3 | 46,883 |
| GAGNNNNNNGATG | 0.981 | 421 | 429 | 99.2 | 6.47 | 74.4 | 41,075 |
| Motifs in X. kozodoii FR71 | |||||||
| GATC | 0.997 | 33,373 | 33,460 | 176.4 | 7.43 | 115.0 | 5,874,492 |
| GGATG | 0.691 | 6281 | 9094 | 53.8 | 2.84 | 118.4 | 242,171 |
| GACCC | 0.936 | 2885 | 3082 | 61.4 | 3.25 | 116.9 | 166,942 |
| Motifs in X. kozodoii FR74 | |||||||
| GATC | 0.998 | 33,548 | 33,600 | 185.1 | 7.48 | 120.0 | 6,202,691 |
| GACCC | 0.953 | 2813 | 2953 | 64.4 | 3.22 | 124.7 | 173,373 |
| TTCANNNNNNNGTG/ | 0.998 | 624 | 625 | 155.6 | 7.02 | 120.9 | 96,959 |
| CACNNNNNNNTGAA 2 | 1.000 | 625 | 625 | 138.4 | 5.81 | 119.7 | 79,103 |
| CATCNNNNNNCTC/ | 0.993 | 416 | 419 | 169.0 | 7.18 | 117.1 | 69,857 |
| GAGNNNNNNGATG | 0.988 | 414 | 419 | 147.2 | 6.48 | 117.0 | 60,273 |
| Strain | Tested Phenotypes a | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Btb Adsorption b | Antibiotic Production c | Sheep Blood Hemolysis d | Motility e | Lipolysis of f | |||||
| Tween 20 | Tween 40 | Tween 60 | Tween 80 | Tween 85 | |||||
| X. nematophila F1 WT | B | + | + | ++ | + | + | + | + | - |
| X. nematophila F1+pBBMCS-5 | B | + | + | ++ | + | + | + | + | - |
| X. nematophila F1+pBB-dam | B | + | w | + | + | + | + | + | - |
| Strain or Plasmid | Relevant Genotype and Characteristics | Reference or Source |
|---|---|---|
| Strains | ||
| Xenorhabdus nematophila F1 | Wild type | [60] |
| Xenorhabdus kozodoii FR48 | Wild type | Laboratory collection |
| Xenorhabdus kozodoii FR71 | Wild type | Laboratory collection |
| Xenorhabdus kozodoii FR74 | Wild type | Laboratory collection |
| Escherichia coli WM3064 | thrB1004 pro thi rpsl hsdS lacZΔM15 RP4-1360Δ(araBAD)567 ΔdapA1341::[erm pir (wt)] | [61] |
| E. coli MG1655 | Wild type | [62] |
| Micrococcus luteus | Wild type | Pasteur Institute Culture collection, Paris, France |
| Plasmids | ||
| pBBR1MCS-5 | Cloning vector, GmR | [63] |
| pBB-Dam | 905 pb PCR fragment (dam gene) inserted between EcoRI and BamHI site of pBBR1_MCS5 plasmid | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ginibre, N.; Legrand, L.; Bientz, V.; Ogier, J.-C.; Lanois, A.; Pages, S.; Brillard, J. Diverse Roles for a Conserved DNA-Methyltransferase in the Entomopathogenic Bacterium Xenorhabdus. Int. J. Mol. Sci. 2022, 23, 11981. https://doi.org/10.3390/ijms231911981
Ginibre N, Legrand L, Bientz V, Ogier J-C, Lanois A, Pages S, Brillard J. Diverse Roles for a Conserved DNA-Methyltransferase in the Entomopathogenic Bacterium Xenorhabdus. International Journal of Molecular Sciences. 2022; 23(19):11981. https://doi.org/10.3390/ijms231911981
Chicago/Turabian StyleGinibre, Nadège, Ludovic Legrand, Victoria Bientz, Jean-Claude Ogier, Anne Lanois, Sylvie Pages, and Julien Brillard. 2022. "Diverse Roles for a Conserved DNA-Methyltransferase in the Entomopathogenic Bacterium Xenorhabdus" International Journal of Molecular Sciences 23, no. 19: 11981. https://doi.org/10.3390/ijms231911981
APA StyleGinibre, N., Legrand, L., Bientz, V., Ogier, J.-C., Lanois, A., Pages, S., & Brillard, J. (2022). Diverse Roles for a Conserved DNA-Methyltransferase in the Entomopathogenic Bacterium Xenorhabdus. International Journal of Molecular Sciences, 23(19), 11981. https://doi.org/10.3390/ijms231911981