
Catabolism of Hydroxyproline in Vertebrates: Physiology, Evolution, Genetic Diseases and New siRNA Approach for Treatment

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Posttranslational Modification of Proline Residue
3. Role of Pro Hydroxylation in Proteins
3.1. Hyp in Collagen
3.2. Role of Hyp in Oxygen-Sensing
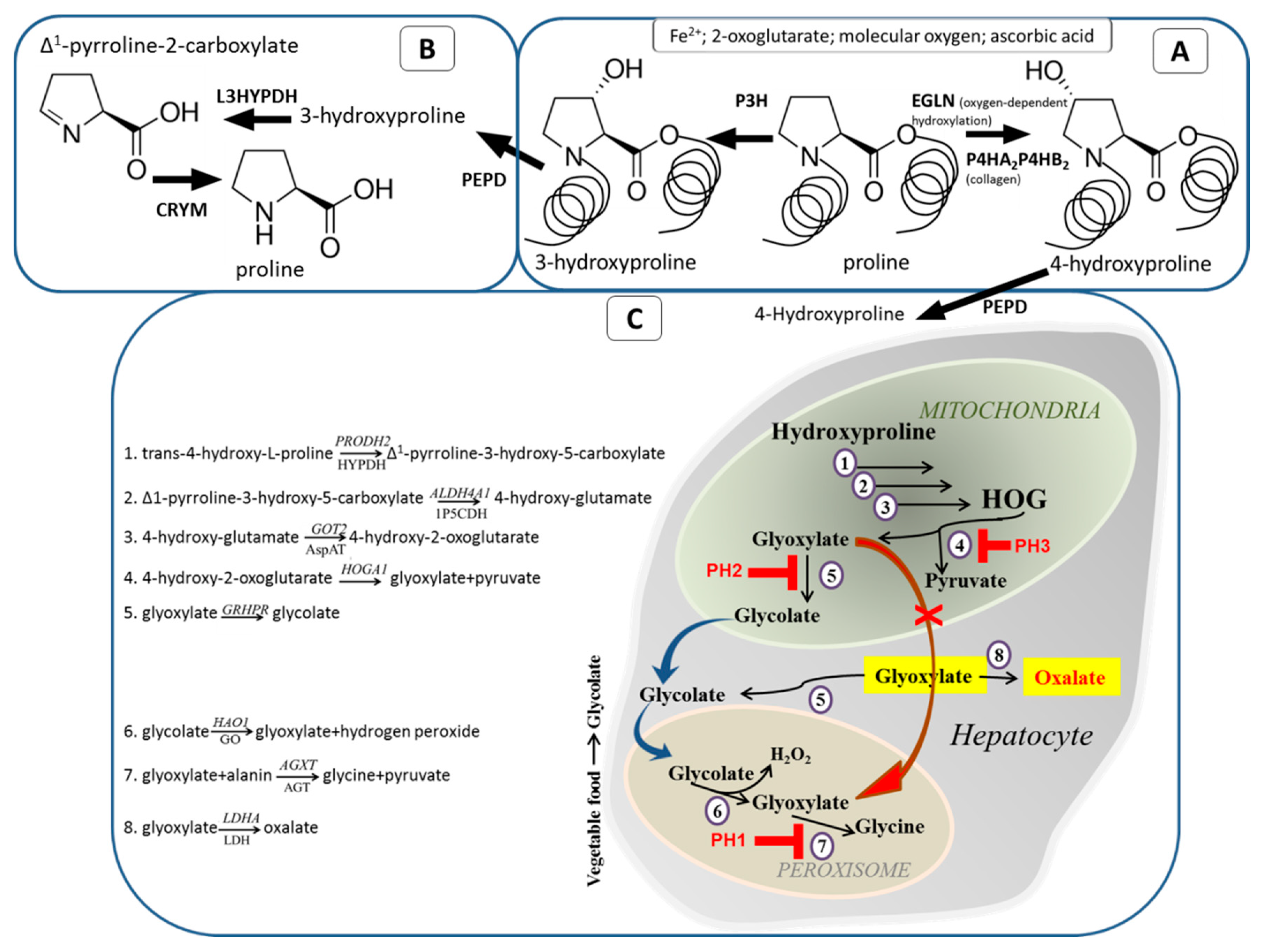
4. Collagen Degradation and Further Metabolism of Hyp
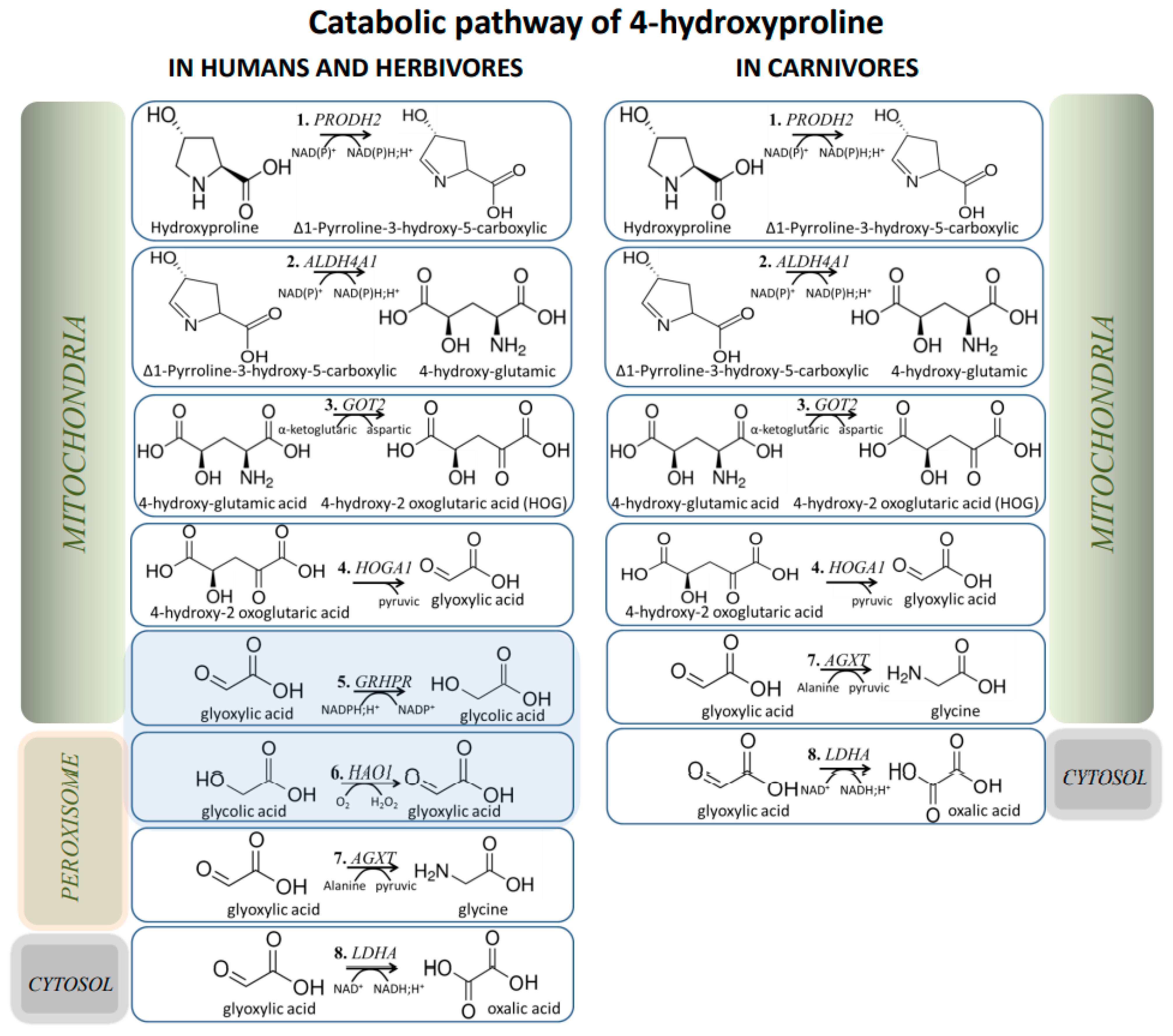
5. Evolutionary Aspects of Hyp/Glyoxylate Pathway
6. Primary Hyperoxaluria and Hydroxyprolinemia
7. PH Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Etherington, D.J.; Pugh, D.; Silver, I.A. Collagen degradation in an experimental inflammatory lesion: Studies on the role of the macrophage. Acta. Biol. Med. Ger. 1981, 40, 1625–1636. [Google Scholar]
- McAnulty, R.J.; Laurent, G.J. Collagen synthesis and degradation in vivo. Evidence for rapid rates of collagen turnover with extensive degradation of newly synthesized collagen in tissues of the adult rat. Coll. Relat. Res. 1987, 7, 93–104. [Google Scholar] [CrossRef]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Rappu, P.; Salo, A.M.; Myllyharju, J.; Heino, J. Role of prolyl hydroxylation in the molecular interactions of collagens. Essays. Biochem. 2019, 63, 325–335. [Google Scholar] [PubMed] [Green Version]
- Buchalski, B.; Wood, K.D.; Challa, A.; Fargue, S.; Holmes, R.P.; Lowther, W.T.; Knight, J. The effects of the inactivation of Hydroxyproline dehydrogenase on urinary oxalate and glycolate excretion in mouse models of primary hyperoxaluria. Biochim. Biophys. Acta Mol. Basis. Dis. 2020, 1866, 165633. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, N.; Aydin, S.; Gillion, V.; Morelle, J.; Jadoul, M. Pathophysiology and Management of Hyperoxaluria and Oxalate Nephropathy: A Review. Am. J. Kidney. Dis. 2021. [Google Scholar] [CrossRef]
- Fargue, S.; Milliner, D.S.; Knight, J.; Olson, J.B.; Lowther, W.T.; Holmes, R.P. Hydroxyproline Metabolism and Oxalate Synthesis in Primary Hyperoxaluria. J. Am. Soc. Nephrol. 2018, 29, 1615–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, J.N.; Wood, K.D.; Knight, J.; Assimos, D.G.; Holmes, R.P. Glyoxal formation and its role in endogenous oxalate synthesis. Adv. Urol. 2012, 2012, 819202. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fargue, S.; Challa, A.K.; Poore, W.; Knight, J.; Wood, K.D. Generation of a GLO-2 deficient mouse reveals its effects on liver carbonyl and glutathione levels. Biochem. Biophys. Rep. 2021, 28, 101138. [Google Scholar] [CrossRef]
- Oatey, P.B.; Lumb, M.J.; Danpure, C.J. Molecular basis of the variable mitochondrial and peroxisomal localisation of alanine-glyoxylate aminotransferase. Eur. J. Biochem. 1996, 241, 374–385. [Google Scholar] [CrossRef]
- Ben-Shalom, E.; Frishberg, Y. Primary hyperoxalurias: Diagnosis and treatment. Pediatr. Nephrol. 2015, 30, 1781–1791. [Google Scholar] [CrossRef]
- Sas, D.J.; Harris, P.C.; Milliner, D.S. Recent advances in the identification and management of inherited hyperoxalurias. Urolithiasis 2019, 47, 79–89. [Google Scholar] [CrossRef]
- Belostotsky, R.; Frishberg, Y. Novel therapeutic approaches for the primary hyperoxalurias. Pediatr. Nephrol. 2021, 36, 2593–2606. [Google Scholar] [CrossRef] [PubMed]
- Frishberg, Y.; Deschênes, G.; Groothoff, J.W.; Hulton, S.A.; Magen, D.; Harambat, J.; Van’t Hoff, W.G.; Lorch, U.; Milliner, D.S.; Lieske, J.C.; et al. Phase 1/2 Study of Lumasiran for Treatment of Primary Hyperoxaluria Type 1: A Placebo-Controlled Randomized Clinical Trial. Clin. J. Am. Soc. Nephrol. 2021, 16, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; Van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.E.; Nagel, M.B.; Dziomba, S.; Merkher, Y.; Sivan, S.S.; Heinz, A. Prolyl hydroxylation in elastin is not random. Biochim. Biophys. Acta 2016, 1860, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.H.; Ongusaha, P.P.; Myllyharju, J.; Cheng, D.; Pakkanen, O.; Shi, Y.; Lee, S.W.; Peng, J.; Shi, Y. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature 2008, 455, 421–424. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Gjaltema, R.A.; Bank, R.A. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Kajihara, D.; Hon, C.C.; Abdullah, A.N.; Wosniak, J., Jr.; Moretti, A.I.S.; Poloni, J.F.; Bonatto, D.; Hashimoto, K.; Carninci, P.; Laurindo, F.R.M. Analysis of splice variants of the human protein disulfide isomerase (P4HB) gene. BMC Genom. 2020, 21, 766. [Google Scholar] [CrossRef] [PubMed]
- Soares Moretti, A.I.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 2017, 617, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.M.; Eyre, D.R. Collagen prolyl 3-hydroxylation: A major role for a minor post-translational modification? Connect. Tissue. Res. 2013, 54, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, S.M.; Besio, R.; Heard-Lipsmeyer, M.E.; Dimori, M.; Castagnola, P.; Swain, F.L.; Gaddy, D.; Diekman, A.B.; Morello, R. Expression characterization and functional implication of the collagen-modifying Leprecan proteins in mouse gonadal tissue and mature sperm. AIMS Genet. 2018, 5, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Garibaldi, N.; Leoni, L.; Cipolla, L.; Sabbioneda, S.; Biggiogera, M.; Mottes, M.; Aglan, M.; Otaify, G.A.; Temtamy, S.A.; et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis. Model. Mech. 2019, 12, dmm038521. [Google Scholar] [CrossRef] [Green Version]
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Ko, A.; Oh, Y.T.; Shi, P.; D’Angelo, F.; Frangaj, B.; Koller, A.; Chen, E.I.; Cardozo, T.; Iavarone, A.; et al. Proline Hydroxylation Primes Protein Kinases for Autophosphorylation and Activation. Mol. Cell 2020, 79, 376–389. [Google Scholar] [CrossRef]
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020, 287, 3888–3906. [Google Scholar] [CrossRef]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, B.; Thiagarajan, G.; Madhan, B.; Kar, K. Triple-helical peptides: An approach to collagen conformation, stability, and self-association. Biopolymers 2008, 89, 345–353. [Google Scholar] [CrossRef]
- Bella, J.; Brodsky, B.; Berman, H.M. Hydration structure of a collagen peptide. Structure 1995, 3, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Vitagliano, L.; Berisio, R.; Mazzarella, L.; Zagari, A. Structural bases of collagen stabilization induced by Pro hydroxylation. Biopolymers 2001, 58, 459–464. [Google Scholar] [CrossRef]
- Taga, Y.; Tanaka, K.; Hattori, S.; Mizuno, K. In-depth correlation analysis demonstrates that 4-hydroxyproline at the Yaa position of Gly-Xaa-Yaa repeats dominantly stabilizes collagen triple helix. Matrix. Biol. Plus 2021, 10, 100067. [Google Scholar] [CrossRef]
- Hamaia, S.; Farndale, R.W. Integrin recognition motifs in the human collagens. Adv. Exp. Med. Biol. 2014, 819, 127–142. [Google Scholar]
- Chen, E.A.; Lin, Y.S. Using synthetic peptides and recombinant collagen to understand DDR-collagen interactions. Biochim. Biophys. Acta. Mol. Cell. Res. 2019, 1866, 118458. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Ichinohe, T.; Okuma, M.; Farndale, R.W.; Barnes, M.J. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc. Res. 1999, 41, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, N.T.; Zientek, K.D.; Pokidysheva, E.N.; Bächinger, H.P. Post-translational modification of type IV collagen with 3-hydroxyproline affects its interactions with glycoprotein VI and nidogens 1 and 2. J. Biol. Chem. 2018, 293, 5987–5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.C.; Wu, C.Y.; Yang, H.Y. Discoveries of how cells sense oxygen win the 2019 Nobel Prize in Physiology or medicine. Biomed. J. 2020, 43, 434–437. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Singh, A.K.; Carroll, K.; Perkovic, V.; Solomon, S.; Jha, V.; Johansen, K.L.; Lopes, R.D.; Macdougall, I.C.; Obrador, G.T.; Waikar, S.S.; et al. Daprodustat for the Treatment of Anemia in Patients Undergoing Dialysis. N. Engl. J. Med. 2021, 385, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef]
- Eni-Aganga, I.; Lanaghan, Z.M.; Balasubramaniam, M.; Dash, C.; Pandhare, J. PROLIDASE: A Review from Discovery to its Role in Health and Disease. Front. Mol. Biosci. 2021, 8, 849. [Google Scholar] [CrossRef]
- Visser, W.F.; Verhoeven-Duif, N.M.; de Koning, T.J. Identification of a human trans-3-hydroxy-L-proline dehydratase, the first characterized member of a novel family of proline racemase-like enzymes. J. Biol. Chem. 2012, 287, 21654–21662. [Google Scholar] [CrossRef] [Green Version]
- Hallen, A.; Cooper, A.J.; Jamie, J.F.; Karuso, P. Insights into Enzyme Catalysis and Thyroid Hormone Regulation of Cerebral Ketimine Reductase/μ-Crystallin Under Physiological Conditions. Neurochem. Res. 2015, 40, 1252–1266. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.; Frank, L. Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 1980, 49, 1005–1061. [Google Scholar] [CrossRef]
- Knight, J.; Jiang, J.; Assimos, D.G.; Holmes, R.P. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney. Int. 2006, 70, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summitt, C.B.; Johnson, L.C.; Jönsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; He, W.; Wu, G. Hydroxyproline in animal metabolism, nutrition, and cell signaling. Amino Acids 2021. [Google Scholar] [CrossRef]
- Monico, C.G.; Rossetti, S.; Belostotsky, R.; Cogal, A.G.; Herges, R.M.; Seide, B.M.; Olson, J.B.; Bergstrahl, E.J.; Williams, H.J.; Haley, W.E.; et al. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin. J. Am. Soc. Nephrol. 2011, 6, 2289–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belostotsky, R.; Pitt, J.J.; Frishberg, Y. Primary hyperoxaluria type III—A model for studying perturbations in glyoxylate metabolism. J. Mol. Med. 2012, 90, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [Green Version]
- Birdsey, G.M.; Lewin, J.; Holbrook, J.D.; Simpson, V.R.; Cunningham, A.A.; Danpure, C.J. A comparative analysis of the evolutionary relationship between diet and enzyme targeting in bats, marsupials and other mammals. Proc. Biol. Sci. 2005, 272, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, A.; Xue, H.H.; Oda, T.; Uchida, C.; Sugiyama, T.; Maeda-Nakai, E.; Sato, K.; Nagai, E.; Watanabe, S.; Takayama, T. Oxalate synthesis in mammals: Properties and subcellular distribution of serine:pyruvate/alanine:glyoxylate aminotransferase in the liver. Mol. Urol. 2000, 4, 333–340. [Google Scholar] [PubMed]
- Danpure, C.J. Variable peroxisomal and mitochondrial targeting of alanine: Glyoxylate aminotransferase in mammalian evolution and disease. Bioessays 1997, 19, 317–326. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, H.; Yuan, X.; Rossiter, S.J.; Zhang, S. Multiple adaptive losses of alanine-glyoxylate aminotransferase mitochondrial targeting in fruit-eating bats. Mol. Biol. Evol. 2012, 29, 1507–1511. [Google Scholar] [CrossRef] [Green Version]
- Holbrook, J.D.; Birdsey, G.M.; Yang, Z.; Bruford, M.W.; Danpure, C.J. Molecular adaptation of alanine:glyoxylate aminotransferase targeting in primates. Mol. Biol. Evol. 2000, 17, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.J.; Xia, J.M.; Wang, Q.; Yu, J.L.; Song, Z.; Zhao, H. Diet and Adaptive Evolution of Alanine-Glyoxylate Aminotransferase Mitochondrial Targeting in Birds. Mol. Biol. Evol. 2020, 37, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, A. Studies on a unique organelle localization of a liver enzyme, serine:pyruvate (or alanine:glyoxylate) aminotransferase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.; López-Ojén, M.; Funcasta-Calderón, R.; Ameneiros-Rodríguez, E.; Donapetry-García, C.; Vila-Altesor, M.; Rodríguez-Seijas, J. Comprehensive review on lactate metabolism in human health. Mitochondrion 2014, 17, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Iepsen, U.W.; Plovsing, R.R.; Tjelle, K.; Foss, N.B.; Meyhoff, C.S.; Ryrsø, C.K.; Berg, R.M.G.; Secher, N.H. The role of lactate in sepsis and COVID-19: Perspective from contracting skeletal muscle metabolism. Exp. Physiol. 2021, 31. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.J.; Knight, J.; Murray, M.S.; Milliner, D.S.; Holmes, R.P.; Lowther, W. T: 4-Hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. Biochim. Biophys. Acta 2012, 1822, 1544–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Burke, J.; Bunker, R.D.; Mok, Y.F.; Griffin, M.D.; Baker, E.N.; Loomes, K.M. Regulation of human 4-hydroxy-2-oxoglutarate aldolase by pyruvate and α-ketoglutarate: Implications for primary hyperoxaluria type-3. Biochem. J. 2019, 476, 3369–3383. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, R.; Kivirikko, K.I. Hydroxyprolinemia: An apparently harmless familial metabolic disorder. N. Engl. J. Med. 1970, 283, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Staufner, C.; Haack, T.B.; Feyh, P.; Gramer, G.; Raga, D.E.; Terrile, C.; Sauer, S.; Okun, J.G.; Fang-Hoffmann, J.; Mayatepek, E.; et al. Genetic cause and prevalence of hydroxyprolinemia. J. Inherit. Metab. Dis. 2016, 39, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Frishberg, Y.; Zeharia, A.; Lyakhovetsky, R.; Bargal, R.; Belostotsky, R. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J. Med. Genet. 2014, 51, 526–529. [Google Scholar] [CrossRef] [PubMed]
- McGregor, T.L.; Hunt, K.A.; Yee, E.; Mason, D.; Nioi, P.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Lawlor, D.A.; et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. eLife 2020, 9, e54363. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Kuhn, H.; Kohbrok, S.; Volland, R.; Franklin, J.; Hero, B.; Beck, B.B.; Hoppe, B. Vitamin B6 in primary hyperoxaluria I: First prospective trial after 40 years of practice. Clin. J. Am. Soc. Nephrol. 2014, 9, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, E.C.; Lieske, J.C.; Seide, B.M.; Olson, J.B.; Mehta, R.; Milliner, D.S. Recovery From Dialysis in Patients With Primary Hyperoxaluria Type 1 Treated With Pyridoxine: A Report of 3 Cases. Am. J. Kidney Dis. 2021, 77, 816–819. [Google Scholar] [CrossRef]
- Forbes, T.A.; Brown, B.D.; Lai, C. Therapeutic RNA interference: A novel approach to the treatment of primary hyperoxaluria. Br. J. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Moya-Garzon, M.D.; Gomez-Vidal, J.A.; Alejo-Armijo, A.; Altarejos, J.; Rodriguez-Madoz, J.R.; Fernandes, M.X.; Salido, E.; Salido, S.; Diaz-Gavilan, M. Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias. J. Pers. Med. 2021, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Martin-Higueras, C.; Hoppe, B. Novel therapeutic approaches in primary hyperoxaluria. Expert. Opin. Emerg. Drugs. 2018, 23, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Titze-de-Almeida, R.; David, C.; Titze-de-Almeida, S.S. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm. Res. 2017, 34, 1339–1363. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Keam, S.J. Lumasiran: First Approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Ariceta, G.; Barrios, K.; Brown, B.D.; Hoppe, B.; Rosskamp, R.; Langman, C.B. Hepatic Lactate Dehydrogenase A: An RNA Interference Target for the Treatment of All Known Types of Primary Hyperoxaluria. Kidney Int. Rep. 2021, 6, 1088–1098. [Google Scholar] [CrossRef]
- Lai, C.; Pursell, N.; Gierut, J.; Saxena, U.; Zhou, W.; Dills, M.; Diwanji, R.; Dutta, C.; Koser, M.; Nazef, N.; et al. Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Mol. Ther. 2018, 26, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, B.; Koch, A.; Cochat, P.; Garrelfs, S.F.; Baum, M.A.; Groothoff, J.W.; Lipkin, G.; Coenen, M.; Schalk, G.; Amrite, A.; et al. Safety, pharmacodynamics, and exposure-response modeling results from a first in human phase 1 study of nedosiran in primary hyperoxaluria. Kidney Int. 2021. [Google Scholar] [CrossRef]
- Shee, K.; Ahn, J.; Hamouche, F.; Mena, J.; Chi, T.; Stoller, M.L. Nedosiran Dramatically Reduces Serum Oxalate in Dialysis-Dependent Primary Hyperoxaluria 1: A Compassionate Use Case Report. Urology 2021, 156, e147–e149. [Google Scholar] [CrossRef]
- Baum, M.A.; Langman, C.B.; Cochat, P.; Lieske, J.; Moochhala, S.; Hamamoto, S.; Satoh, H.; Mourani, C.; Ariceta, G.; Amrite, A.; et al. PHYOX2: Nedosiran reduced urinary oxalate excretion in patients with primary hyperoxaluria. J. Am. Soc. Nephrol. 2021, 32. Available online: https://www.asn-online.org/education/kidneyweek/2021/program-abstract.aspx?controlId=3627285 (accessed on 21 December 2021).
- Dicerna Pharmaceuticals, Press Release 19 October 2021. Available online: https://investors.dicerna.com/news-releases/news-release-details/dicerna-announces-results-phyoxtm4-single-dose-study-nedosiran (accessed on 21 December 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belostotsky, R.; Frishberg, Y. Catabolism of Hydroxyproline in Vertebrates: Physiology, Evolution, Genetic Diseases and New siRNA Approach for Treatment. Int. J. Mol. Sci. 2022, 23, 1005. https://doi.org/10.3390/ijms23021005
Belostotsky R, Frishberg Y. Catabolism of Hydroxyproline in Vertebrates: Physiology, Evolution, Genetic Diseases and New siRNA Approach for Treatment. International Journal of Molecular Sciences. 2022; 23(2):1005. https://doi.org/10.3390/ijms23021005
Chicago/Turabian StyleBelostotsky, Ruth, and Yaacov Frishberg. 2022. "Catabolism of Hydroxyproline in Vertebrates: Physiology, Evolution, Genetic Diseases and New siRNA Approach for Treatment" International Journal of Molecular Sciences 23, no. 2: 1005. https://doi.org/10.3390/ijms23021005
APA StyleBelostotsky, R., & Frishberg, Y. (2022). Catabolism of Hydroxyproline in Vertebrates: Physiology, Evolution, Genetic Diseases and New siRNA Approach for Treatment. International Journal of Molecular Sciences, 23(2), 1005. https://doi.org/10.3390/ijms23021005