
Examination of the Novel Sigma-1 Receptor Antagonist, SI 1/28, for Antinociceptive and Anti-allodynic Efficacy against Multiple Types of Nociception with Fewer Liabilities of Use


 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
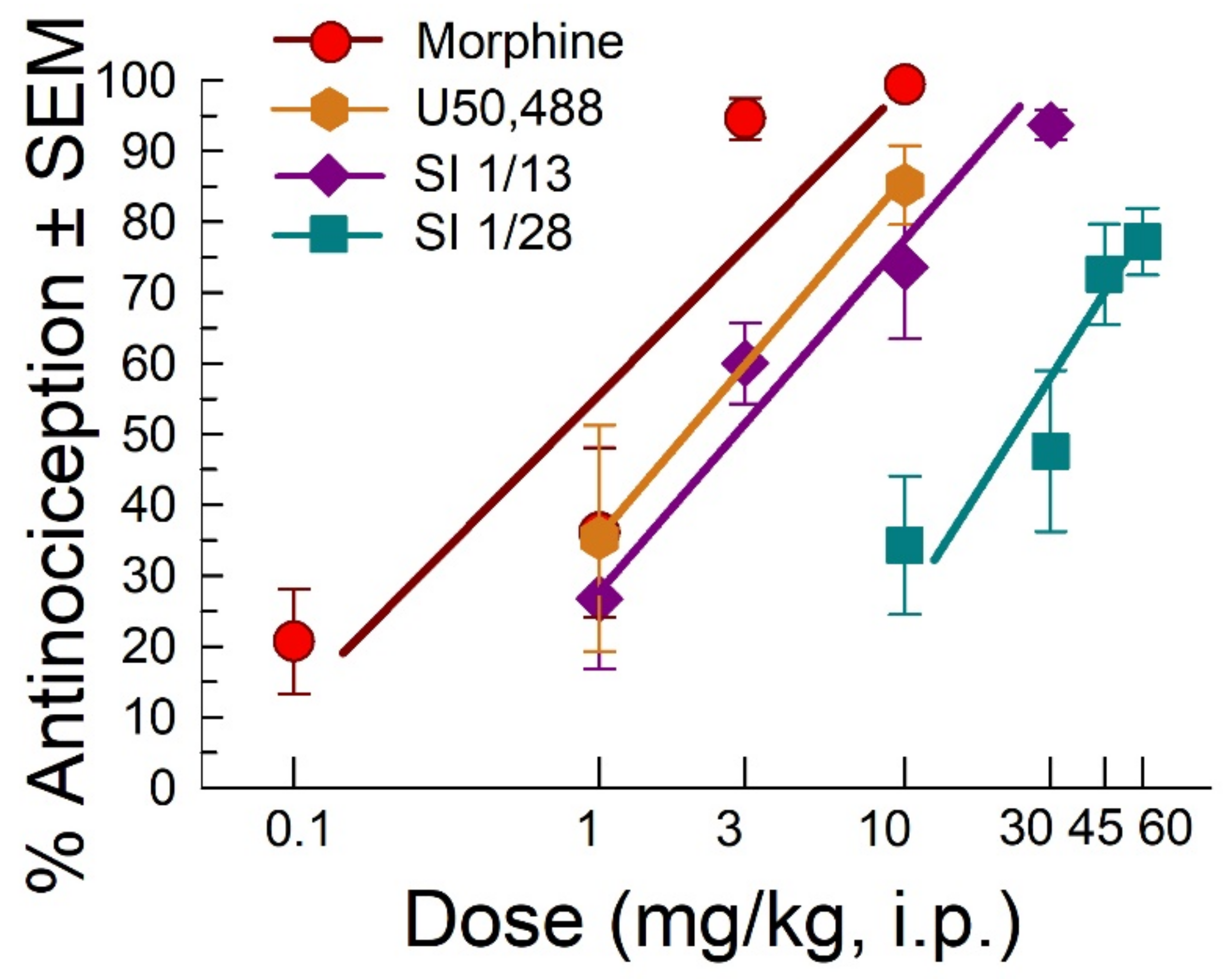
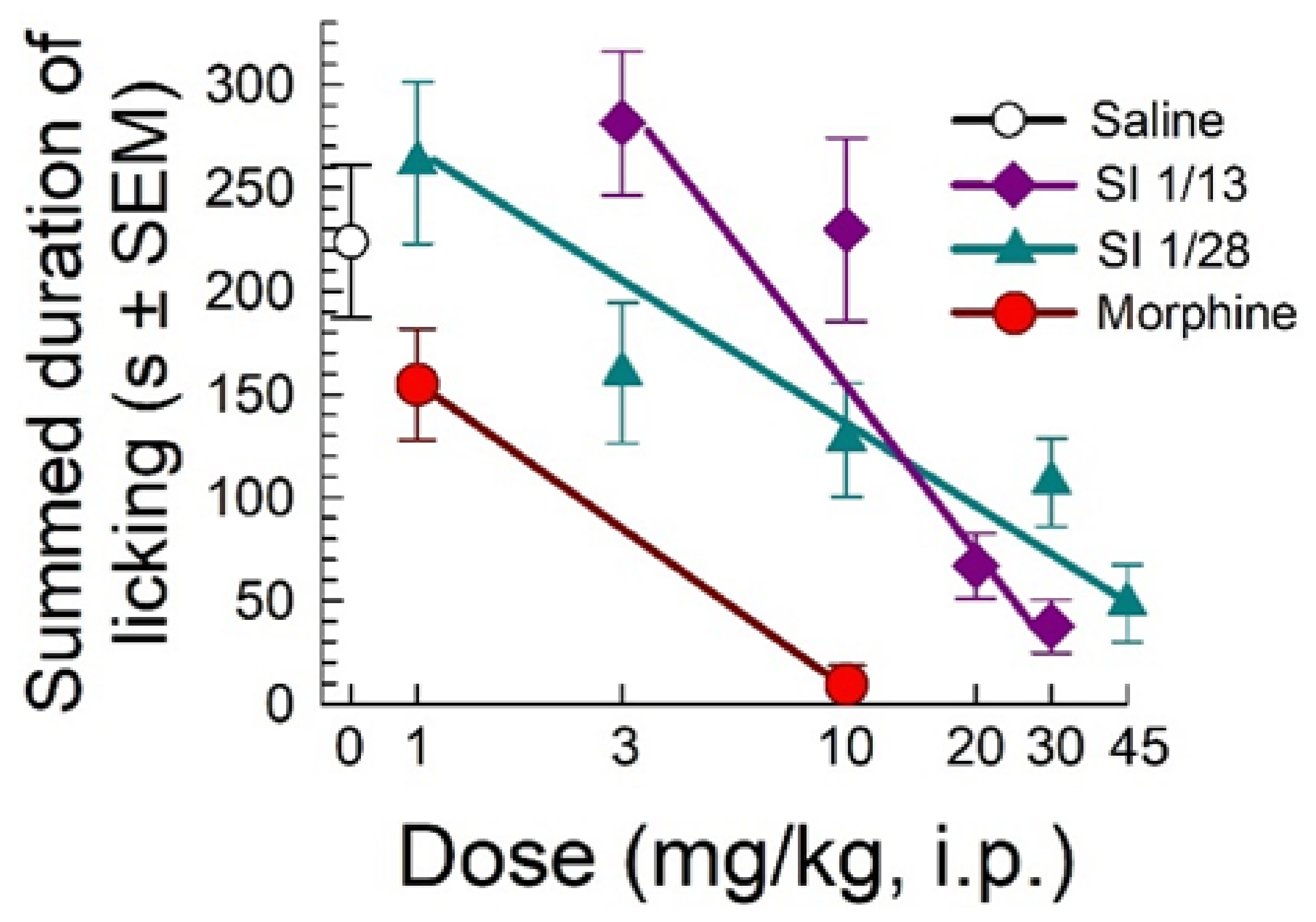
2.1. SI 1/28 Induces Dose-Dependent Antinociception in Mouse Models of Visceral Chemical and Inflammatory Nociception
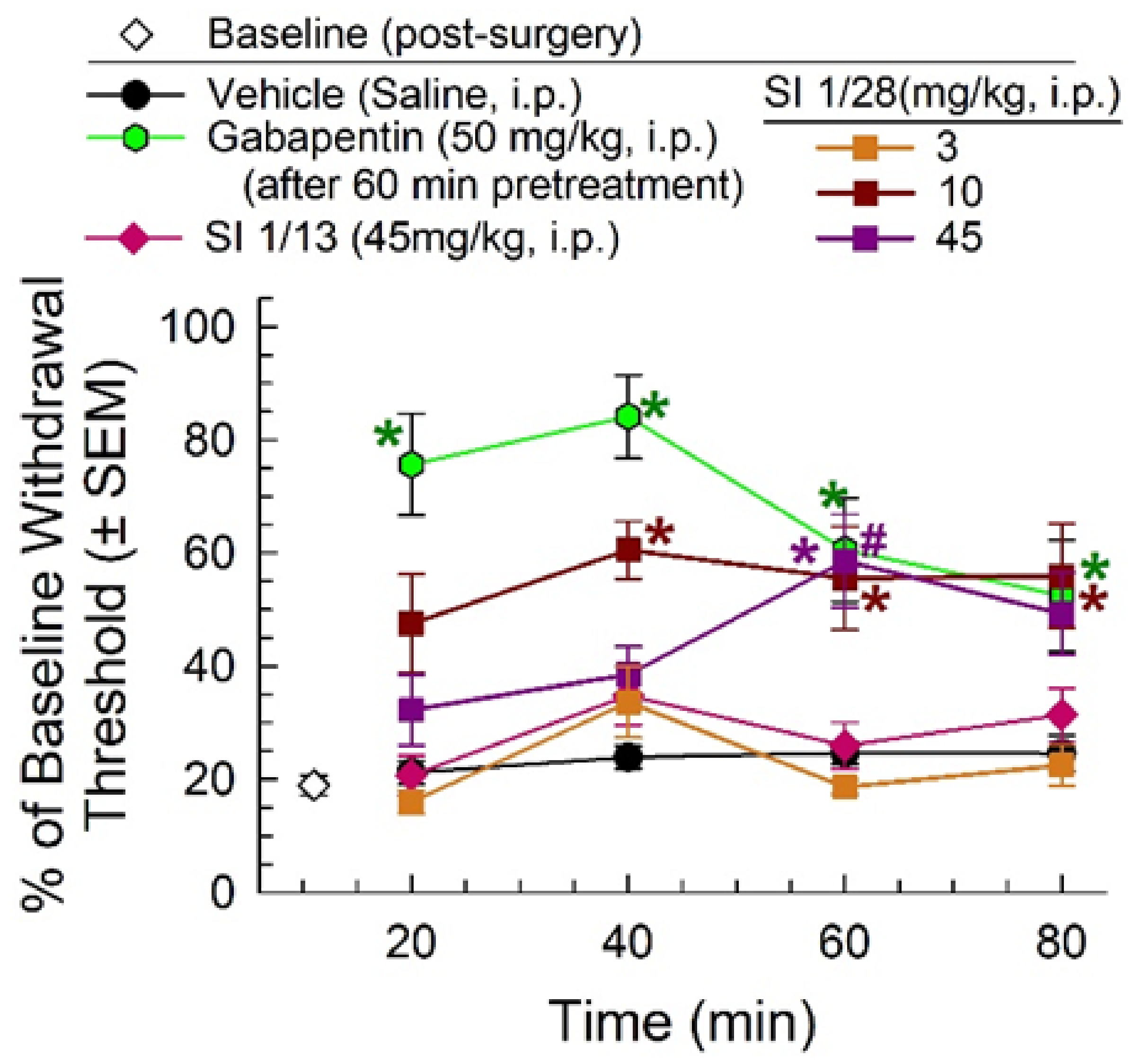
2.2. Anti-Allodynic Effects of SI 1/28
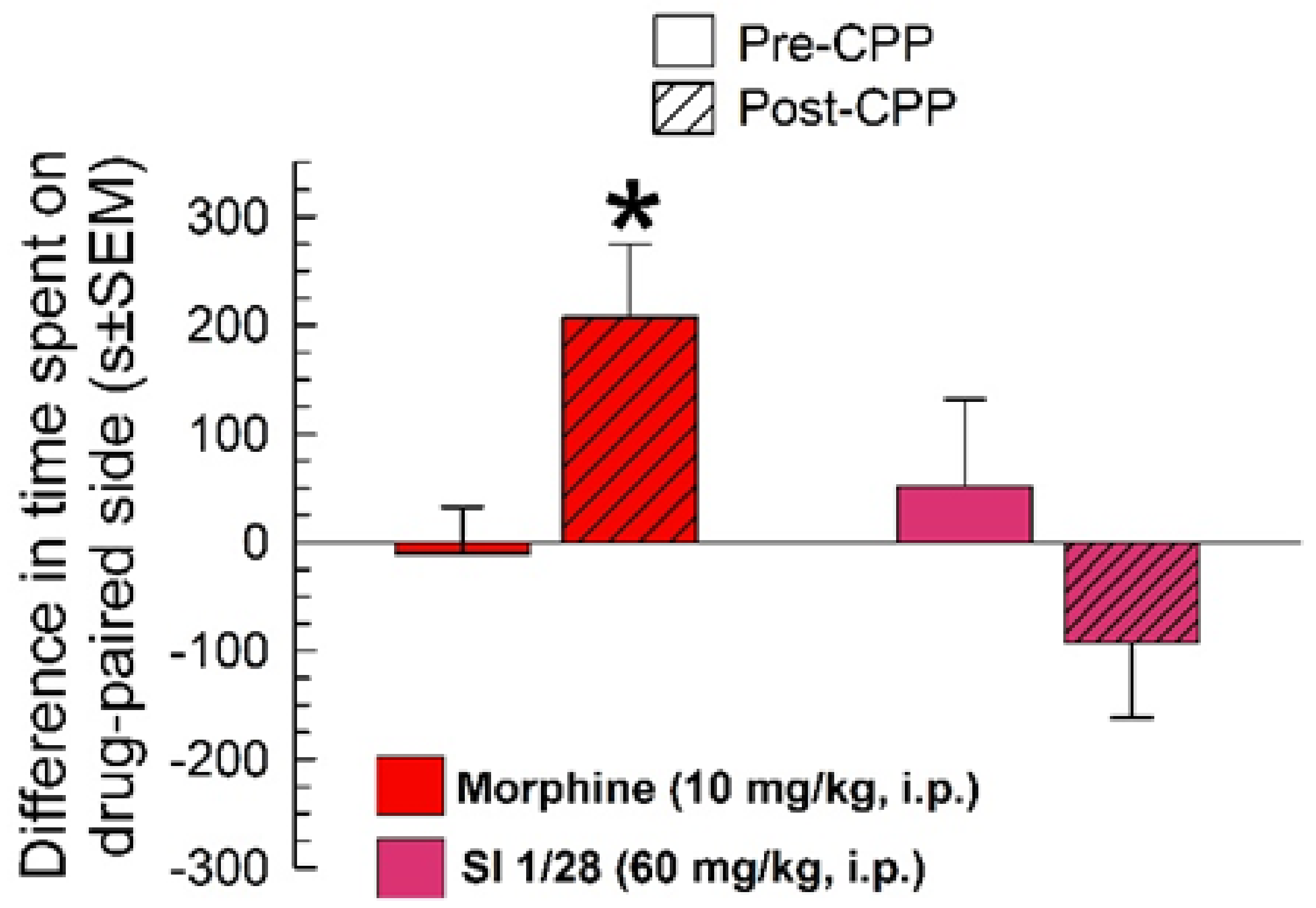
2.3. Evaluation of SI 1/28 for Potential Clinical Liabilities
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Materials, Drug Preparation, and Administration
4.3. Behavioral Assays
4.3.1. Acetic Acid Stretching Assay
4.3.2. Formalin Assay
4.3.3. Chronic Constriction Injury (CCI)
4.3.4. Conditioned Place Preference (CPP)
4.3.5. Respiratory Depression and Spontaneous Locomotor Testing with CLAMS
4.3.6. Rotarod Assay to Assess Motor Coordination
4.3.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicholson, B.; Verma, S. Comorbidities in chronic neuropathic pain. Pain Med. 2004, 5, S9–S27. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilron, I.; Bailey, J.M.; Tu, D.; Holden, R.R.; Jackson, A.C.; Houlden, R.L. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: A double-blind, randomised controlled crossover trial. Lancet 2009, 374, 1252–1261. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.; Benveniste, H.; McLellan, A.T. Use and misuse of opioids in chronic pain. Annu. Rev. Med. 2018, 69, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boom, M.; Niesters, M.; Sarton, E.; Aarts, L.; Smith, T.W.; Dahan, A. Non-analgesic effects of opioids: Opioid-induced respiratory depression. Curr. Pharm. Des. 2012, 18, 5994–6004. [Google Scholar] [CrossRef]
- Nelson, L.S.; Juurlink, D.N.; Perrone, J. Addressing the opioid epidemic. JAMA 2015, 314, 1453–1454. [Google Scholar] [CrossRef]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.M.; Wang, F.; Qiu, C.; Itson-Zoske, B.; Hogan, Q.H.; Yu, H. Sigma-1 receptor activity in primary sensory neurons is a critical driver of neuropathic pain. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Romero, L.; Merlos, M.; Vela, J.M. Antinociception by Sigma-1 Receptor Antagonists: Central and Peripheral Effects. Adv. Pharmacol. 2016, 75, 179–215. [Google Scholar]
- Merlos, M.; Romero, L.; Zamanillo, D.; Plata-Salamán, C.; Vela, J.M. Sigma-1 Receptor and Pain. Handb. Exp. Pharmacol. 2017, 244, 131–161. [Google Scholar] [PubMed]
- Castany, S.; Gris, G.; Vela, J.M.; Verdú, E.; Boadas-Vaello, P. Critical role of sigma-1 receptors in central neuropathic pain-related behaviours after mild spinal cord injury in mice. Sci. Rep. 2018, 8, 3873. [Google Scholar] [CrossRef]
- Zamanillo, D.; Romero, L.; Merlos, M.; Vela, J.M. Sigma 1 receptor: A new therapeutic target for pain. Eur. J. Pharmacol. 2013, 716, 78–93. [Google Scholar] [CrossRef]
- Gris, G.; Merlos, M.; Vela, J.M.; Zamanillo, D.; Portillo-Salido, E. S1RA, a selective sigma-1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund’s adjuvant models in mice. Behav. Pharmacol. 2014, 25, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Caparrós, I.; Perazzoli, G.; Yeste, S.; Cikes, D.; Baeyens, J.M.; Cobos, E.J.; Nieto, F.R. Sigma-1 receptor inhibition reduces neuropathic pain induced by partial sciatic nerve transection in mice by opioid-dependent and -independent mechanisms. Front. Pharmacol. 2019, 10, 613. [Google Scholar] [CrossRef]
- Bruna, J.; Videla, S.; Argyriou, A.A.; Velasco, R.; Villoria, J.; Santos, C.; Nadal, C.; Cavaletti, G.; Alberti, P.; Briani, C.; et al. Efficacy of a novel sigma-1 receptor antagonist for oxaliplatin-induced neuropathy: A randomized, double-blind, placebo-controlled phase IIa clinical trial. Neurotherapeutics 2018, 15, 178–189. [Google Scholar] [CrossRef]
- James, M.L.; Shen, B.; Zavaleta, C.L.; Nielsen, C.H.; Mesangeau, C.; Vuppala, P.K.; Chan, C.; Avery, B.A.; Fishback, J.A.; Matsumoto, R.R.; et al. New positron emission tomography (PET) radioligand for imaging σ-1 receptors in living subjects. J. Med. Chem. 2012, 55, 8272–8282. [Google Scholar] [CrossRef]
- James, M.L.; Shen, B.; Nielsen, C.H.; Behera, D.; Buckmaster, C.L.; Mesangeau, C.; Zavaleta, C.; Vuppala, P.K.; Jamalapuram, S.; Avery, B.A.; et al. Evaluation of σ-1 receptor radioligand 18F-FTC-146 in rats and squirrel monkeys using PET. J. Nucl. Med. 2014, 55, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; James, M.L.; Andrews, L.; Lau, C.; Chen, S.; Palner, M.; Miao, Z.; Arksey, N.C.; Shuhendler, A.J.; Scatliffe, S.; et al. Further validation to support clinical translation of [18F]FTC-146 for imaging sigma-1 receptors. EJNMMI Res. 2015, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Park, J.H.; Hjørnevik, T.; Cipriano, P.W.; Yoon, D.; Gulaka, P.K.; Holly, D.; Behera, D.; Avery, B.A.; Gambhir, S.S.; et al. Radiosynthesis and First-In-Human PET/MRI Evaluation with Clinical-Grade [18F]FTC-146. Mol. Imaging Biol. 2017, 19, 779–786. [Google Scholar] [CrossRef]
- Romeo, G.; Bonanno, F.; Wilson, L.L.; Arena, E.; Modica, M.N.; Pittalà, V.; Salerno, L.; Prezzavento, O.; McLaughlin, J.P.; Intagliata, S. Development of new benzylpiperazine derivatives as σ1 receptor ligands with in vivo antinociceptive and anti-allodynic effects. ACS Chem. Neurosci. 2021, 12, 2003–2012. [Google Scholar] [CrossRef]
- Romeo, G.; Ciaffaglione, V.; Amata, E.; Dichiara, M.; Calabrese, L.; Vanella, L.; Sorrenti, V.; Grosso, S.; D’Amico, A.G.; D’Agata, V.; et al. Combination of heme oxygenase-1 inhibition and sigma receptor modulation for anticancer activity. Molecules 2021, 26, 3860. [Google Scholar] [CrossRef]
- Fallica, A.N.; Pittalà, V.; Modica, M.N.; Salerno, L.; Romeo, G.; Marrazzo, A.; Helal, M.A.; Intagliata, S. Recent advances in the development of sigma receptor ligands as cytotoxic agents: A medicinal chemistry perspective. J. Med. Chem. 2021, 64, 7926–7962. [Google Scholar] [CrossRef]
- Schepmann, D.; Neue, C.; Westphälinger, S.; Müller, C.; Bracher, F.; Lange, C.; Bednarski, P.; Almansa, C.; Friedland, K.; Räbiger, V.; et al. Pharmacological characterization of high-affinity σ1 receptor ligands with spirocyclic thienopyran and thienofuran scaffold. J. Pharm. Pharmacol. 2020, 72, 236–248. [Google Scholar] [CrossRef] [Green Version]
- Cirino, T.J.; Eans, S.O.; Medina, J.M.; Wilson, L.L.; Mottinelli, M.; Intagliata, S.; McCurdy, C.R.; McLaughlin, J.P. Characterization of sigma 1 receptor antagonist CM-304 and its analog, AZ-66: Novel therapeutics against allodynia and induced pain. Front. Pharmacol. 2019, 10, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romieu, P.; Meunier, J.; Garcia, D.; Zozime, N.; Martin-Fardon, R.; Bowen, W.D.; Maurice, T. The sigma1 (sigma1) receptor activation is a key step for the reactivation of cocaine conditioned place preference by drug priming. Psychopharmacology 2004, 175, 154–162. [Google Scholar] [CrossRef]
- Chen, S.L.; Hsu, K.Y.; Huang, E.Y.; Lu, R.B.; Tao, P.L. Low doses of dextromethorphan attenuate morphine-induced rewarding via the sigma-1 receptor at ventral tegmental area in rats. Drug Alcohol Depend. 2011, 117, 164–169. [Google Scholar] [CrossRef]
- Abadias, M.; Escriche, M.; Vaqué, A.; Sust, M.; Encina, G. Safety, tolerability and pharmacokinetics of single and multiple doses of a novel sigma-1 receptor antagonist in three randomized phase I studies. Br. J. Clin. Pharmacol. 2013, 75, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Fraser, M.R.; Levine, M. A Comprehensive Approach to Addressing the Opioid Crisis. In A Public Health Guide to Ending the Opioid Epidemic; Oxford University Press: Oxford, UK, 2019. [Google Scholar]
- Merlos, M.; Burgueño, J.; Portillo-Salido, E.; Plata-Salamán, C.R.; Vela, J.M. Pharmacological modulation of the sigma 1 receptor and the treatment of pain. Adv. Exp. Med. Biol. 2017, 964, 85–107. [Google Scholar]
- Vidal-Torres, A.; Fernández-Pastor, B.; Carceller, A.; Vela, J.M.; Merlos, M.; Zamanillo, D. Effects of the selective sigma-1 receptor antagonist S1RA on formalin-induced pain behavior and neurotransmitter release in the spinal cord in rats. J. Neurochem. 2014, 129, 484–494. [Google Scholar] [CrossRef] [Green Version]
- McCall, W.D.; Tanner, K.D.; Levine, J.D. Formalin induces biphasic activity in C-fibers in the rat. Neurosci. Lett. 1996, 208, 45–48. [Google Scholar] [CrossRef]
- Wheeler-Aceto, H.; Cowan, A. Standardization of the rat paw formalin test for the evaluation of analgesics. Psychopharmacology 1991, 104, 35–44. [Google Scholar] [CrossRef]
- Abbott, F.V.; Franklin, K.B.J.; Westbrook, F.R. The formalin test: Scoring properties of the first and second phases of the pain response in rats. Pain 1995, 60, 91–102. [Google Scholar] [CrossRef]
- Baba, H.; Doubell, T.P.; Woolf, C.J. Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J. Neurosci. 1999, 19, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Drews, E.; Zimmer, A. Central sensitization needs sigma receptors. Pain 2009, 145, 269–270. [Google Scholar] [CrossRef]
- Cervero, F.; Laird, J.M.A. Spinal Mechanisms of Visceral Pain and Hyperalgesia. In Synaptic Plasticity in Pain; Malcangio, M., Ed.; Springer: New York, NY, USA, 2009; pp. 289–306. [Google Scholar]
- Puente, B.; Nadal, X.; Portillo-Salido, E.; Sánchez-Arroyos, R.; Ovalle, S.; Palacios, G.; Muro, A.; Romero, L.; Entrena, J.M.; Baeyens, J.M.; et al. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain 2009, 145, 294–303. [Google Scholar] [CrossRef]
- Budd, K. The use of partial antagonist analgesics in the treatment of acute and chronic pain. Can. Anaesth. Soc. J. 1985, 32, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.H.; Knight, A.R.; Woodruff, G.N. [3H]MK-801 labels a site on the N-methyl-D-aspartate receptor channel complex in rat brain membranes. J. Neurochem. 1988, 50, 274–281. [Google Scholar] [CrossRef]
- Palmer, C.P.; Aydar, E.; Jackson, M.B. σ Receptor Modulation of Ion Channels. In Sigma Receptors: Chemistry, Cell Biology and Clinical Implications; Su, T.-P., Matsumoto, R.R., Bowen, W.D., Eds.; Springer: Boston, MA, USA, 2007; pp. 127–149. [Google Scholar]
- Evoy, K.E.; Morrison, M.D.; Saklad, S.R. Abuse and misuse of pregabalin and gabapentin. Drugs 2017, 77, 403–426. [Google Scholar] [CrossRef]
- Manzanedo, C.; Aguilar, M.A.; Miñarro, J. The effects of dopamine D2 and D3 antagonists on spontaneous motor activity and morphine-induced hyperactivity in male mice. Psychopharmacology 1999, 143, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Imam, M.Z.; Kuo, A.; Smith, M.T. Countering opioid-induced respiratory depression by non-opioids that are respiratory stimulants. F1000Research 2020, 9, F1000. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Katz, J.L.; Hiranita, T.; Kopajtic, T.A.; Rice, K.C.; Mesangeau, C.; Narayanan, S.; Abdelazeem, A.H.; McCurdy, C.R. Blockade of cocaine or σ receptor agonist self administration by subtype-selective σ receptor antagonists. J. Pharmacol. Exp. Ther. 2016, 358, 109–124. [Google Scholar] [CrossRef] [Green Version]
- McKendrick, G.; Graziane, N.M. Drug-induced conditioned place preference and its practical use in substance use disorder research. Front. Behav. Neurosci. 2020, 14, 582147. [Google Scholar] [CrossRef]
- Vezina, P.; Stewart, J. Morphine conditioned place preference and locomotion: The effect of confinement during training. Psychopharmacology 1987, 93, 257–260. [Google Scholar] [CrossRef]
- Tapia, M.A.; Lever, J.R.; Lever, S.Z.; Will, M.J.; Park, E.S.; Miller, D.K. Sigma-1 receptor ligand PD144418 and sigma-2 receptor ligand YUN-252 attenuate the stimulant effects of methamphetamine in mice. Psychopharmacology 2019, 236, 3147–3158. [Google Scholar] [CrossRef]
- Maurice, T.; Martin-Fardon, R.; Romieu, P.; Matsumoto, R.R. Sigma1 (σ1) receptor antagonists represent a new strategy against cocaine addiction and toxicity. Neurosci. Biobehav. Rev. 2002, 26, 499–527. [Google Scholar] [CrossRef]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; McLaughlin, J.P.; Houghten, R.A. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Eans, S.O.; Ganno, M.L.; Mizrachi, E.; Houghten, R.A.; Dooley, C.T.; McLaughlin, J.P.; Nefzi, A. Parallel synthesis of hexahydrodiimidazodiazepines heterocyclic peptidomimetics and their in vitro and in vivo activities at μ (MOR), δ (DOR), and κ (KOR) opioid receptors. J. Med. Chem. 2015, 58, 4905–4917. [Google Scholar] [CrossRef]
- Brabant, C.; Quertemont, E.; Tirelli, E. Influence of the dose and the number of drug-context pairings on the magnitude and the long-lasting retention of cocaine-induced conditioned place preference in C57BL/6J mice. Psychopharmacology 2005, 180, 33–40. [Google Scholar] [CrossRef]
- Orsini, C.; Bonito-Oliva, A.; Conversi, D.; Cabib, S. Susceptibility to conditioned place preference induced by addictive drugs in mice of the C57BL/6 and DBA/2 inbred strains. Psychopharmacology 2005, 181, 327–336. [Google Scholar] [CrossRef]
- LaCroix-Fralish, M.L.; Rutkowski, M.D.; Weinstein, J.N.; Mogil, J.S.; Deleo, J.A. The magnitude of mechanical allodynia in a rodent model of lumbar radiculopathy is dependent on strain and sex. Spine 2005, 30, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Mogil, J.S.; Smith, S.B.; O’Reilly, M.K.; Plourde, G. Influence of nociception and stress-induced antinociception on genetic variation in isoflurane anesthetic potency among mouse strains. Anesthesiology 2005, 103, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Feehan, A.K.; Morgenweck, J.; Zhang, X.; Amgott-Kwan, A.T.; Zadina, J.E. Novel endomorphin analogs are more potent and longer-lasting analgesics in neuropathic, inflammatory, postoperative, and visceral pain relative to morphine. J. Pain 2017, 18, 1526–1541. [Google Scholar] [CrossRef] [PubMed]
- Intagliata, S.; Sharma, A.; King, T.I.; Mesangeau, C.; Seminerio, M.; Chin, F.T.; Wilson, L.L.; Matsumoto, R.R.; McLaughlin, J.P.; Avery, B.A.; et al. Discovery of a highly selective sigma-2 receptor ligand, 1-(4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-3-methyl-1H-benzo[d]imidazol-2(3H)-one (CM398), with drug-like properties and antinociceptive effects in vivo. AAPS J. 2020, 22, 94. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthi, I.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Vet. Clin. Pathol. 2012, 41, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Intagliata, S.; Modica, M.N.; Pittalà, V.; Salerno, L.; Siracusa, M.A.; Cagnotto, A.; Salmona, M.; Kurczab, R.; Romeo, G. New N-and O-arylpiperazinylalkyl pyrimidines and 2-methylquinazolines derivatives as 5-HT7 and 5-HT1A receptor ligands: Synthesis, structure-activity relationships, and molecular modeling studies. Bioorganic Med. Chem. 2017, 25, 1250–1259. [Google Scholar] [CrossRef]
- Intagliata, S.; Modica, M.N.; Pittalà, V.; Salerno, L.; Siracusa, M.A.; Cagnotto, A.; Salmona, M.; Romeo, G. Design and synthesis of new homo and hetero bis-piperazinyl-1-propanone derivatives as 5-HT7R selective ligands over 5-HT1AR. Bioorganic Med. Chem. Lett. 2016, 26, 4052–4056. [Google Scholar] [CrossRef]
- Bidlack, J.M.; Cohen, D.J.; McLaughlin, J.P.; Lou, R.; Ye, Y.; Wentland, M.P. 8-Carboxamidocyclazocine: A long-acting, novel benzomorphan. J. Pharmacol. Exp. Ther. 2002, 302, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.Y.; Pitcher, G.M.; Laviolette, S.R.; Whishaw, I.Q.; Tong, K.I.; Kockeritz, L.K.; Wada, T.; Joza, N.A.; Crackower, M.; Goncalves, J.; et al. DREAM is a critical transcriptional repressor for pain modulation. Cell 2002, 108, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Hoot, M.R.; Sim-Selley, L.J.; Poklis, J.L.; Abdullah, R.A.; Scoggins, K.L.; Selley, D.E.; Dewey, W.L. Chronic constriction injury reduces cannabinoid receptor 1 activity in the rostral anterior cingulate cortex of mice. Brain Res. 2010, 1339, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Pitcher, G.M.; Ritchie, J.; Henry, J.L. Nerve constriction in the rat: Model of neuropathic, surgical and central pain. Pain 1999, 83, 37–46. [Google Scholar] [CrossRef]
- Xu, M.; Petraschka, M.; McLaughlin, J.P.; Westenbroek, R.E.; Caron, M.G.; Lefkowitz, R.J.; Czyzyk, T.A.; Pintar, J.E.; Terman, G.W.; Chavkin, C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J. Neurosci. 2004, 24, 4576–4584. [Google Scholar] [CrossRef] [Green Version]
- Gilron, I. Gabapentin and pregabalin for chronic neuropathic and early postsurgical pain: Current evidence and future directions. Curr. Opin. Anaesthesiol. 2007, 20, 456–472. [Google Scholar] [CrossRef]
- Eans, S.O.; Ganno, M.L.; Reilley, K.J.; Patkar, K.A.; Senadheera, S.N.; Aldrich, J.V.; McLaughlin, J.P. The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short-acting κ opioid receptor antagonism in the CNS after oral administration. Br. J. Pharmacol. 2013, 169, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, L.L.; Eans, S.O.; Ramadan-Siraj, I.; Modica, M.N.; Romeo, G.; Intagliata, S.; McLaughlin, J.P. Examination of the Novel Sigma-1 Receptor Antagonist, SI 1/28, for Antinociceptive and Anti-allodynic Efficacy against Multiple Types of Nociception with Fewer Liabilities of Use. Int. J. Mol. Sci. 2022, 23, 615. https://doi.org/10.3390/ijms23020615
Wilson LL, Eans SO, Ramadan-Siraj I, Modica MN, Romeo G, Intagliata S, McLaughlin JP. Examination of the Novel Sigma-1 Receptor Antagonist, SI 1/28, for Antinociceptive and Anti-allodynic Efficacy against Multiple Types of Nociception with Fewer Liabilities of Use. International Journal of Molecular Sciences. 2022; 23(2):615. https://doi.org/10.3390/ijms23020615
Chicago/Turabian StyleWilson, Lisa L., Shainnel O. Eans, Insitar Ramadan-Siraj, Maria N. Modica, Giuseppe Romeo, Sebastiano Intagliata, and Jay P. McLaughlin. 2022. "Examination of the Novel Sigma-1 Receptor Antagonist, SI 1/28, for Antinociceptive and Anti-allodynic Efficacy against Multiple Types of Nociception with Fewer Liabilities of Use" International Journal of Molecular Sciences 23, no. 2: 615. https://doi.org/10.3390/ijms23020615
APA StyleWilson, L. L., Eans, S. O., Ramadan-Siraj, I., Modica, M. N., Romeo, G., Intagliata, S., & McLaughlin, J. P. (2022). Examination of the Novel Sigma-1 Receptor Antagonist, SI 1/28, for Antinociceptive and Anti-allodynic Efficacy against Multiple Types of Nociception with Fewer Liabilities of Use. International Journal of Molecular Sciences, 23(2), 615. https://doi.org/10.3390/ijms23020615