
RNA-Guided AsCas12a- and SpCas9-Catalyzed Knockout and Homology Directed Repair of the Omega-1 Locus of the Human Blood Fluke, Schistosoma mansoni

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
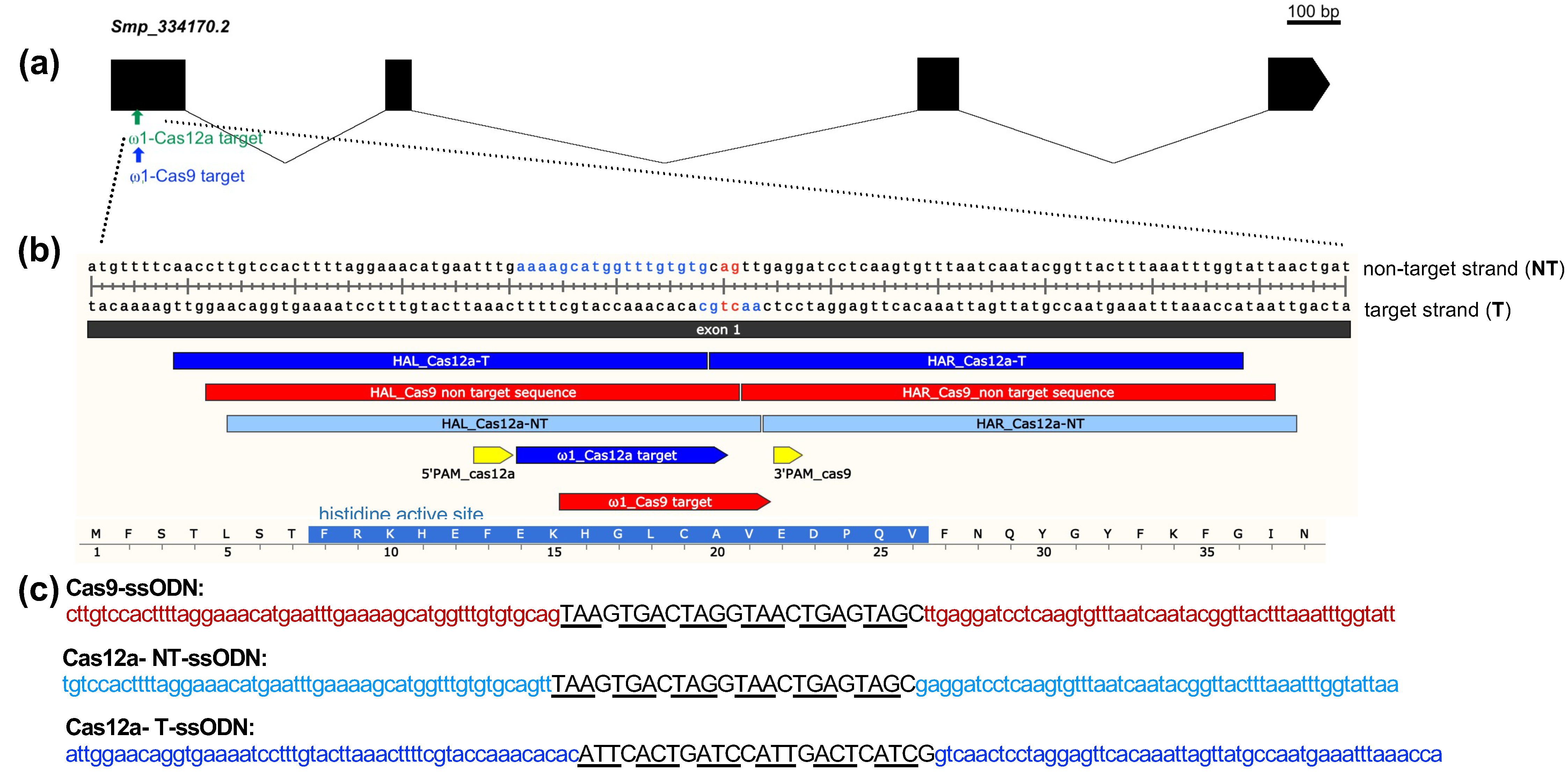
2.1. The Omega-1 Multicopy Gene, Guide RNAs, and Single-Stranded DNA Donor Templates
2.2. Absence of Trans Activity of Activated AsCas12a against the ssODN Donors
2.3. Quantification of Transgene Copy Number by External Standard Curve
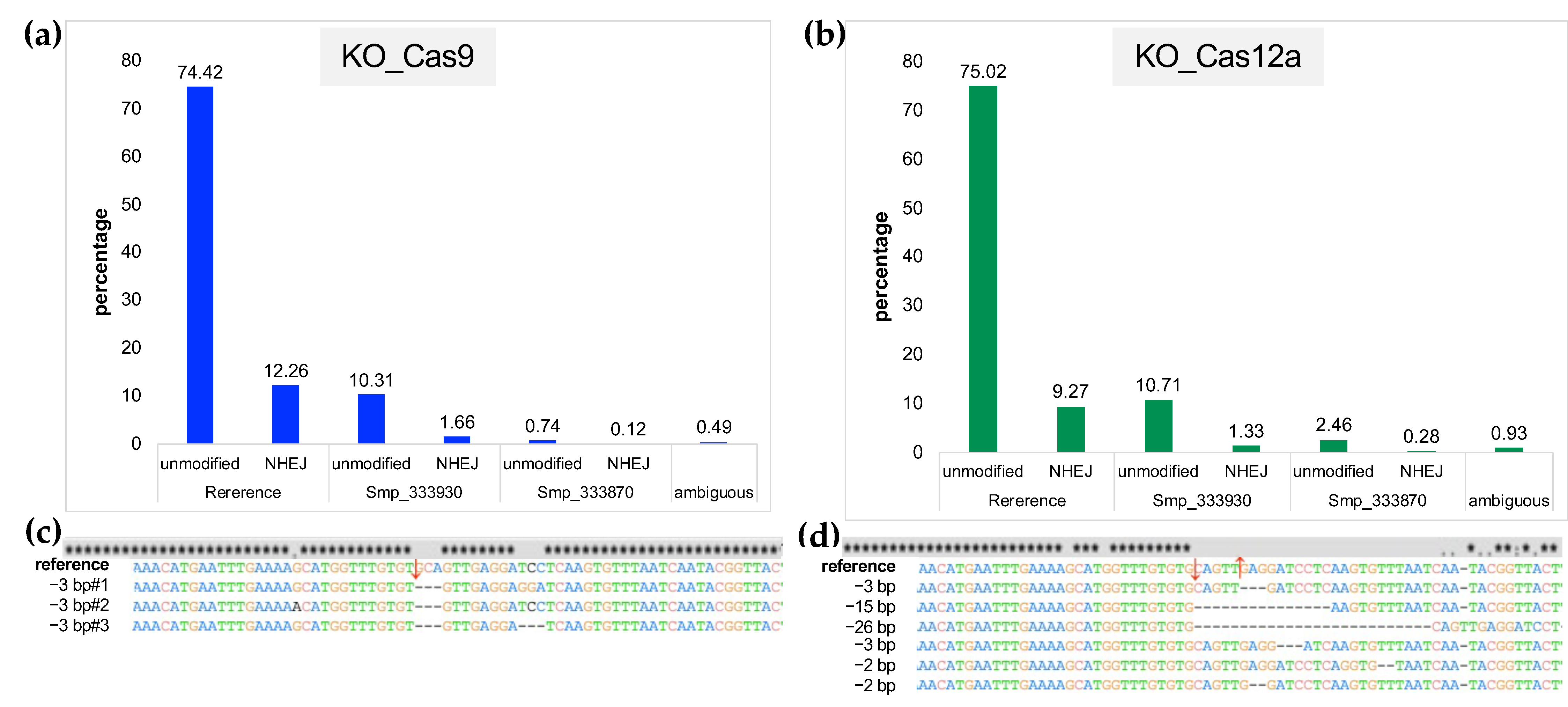
2.4. Estimation of CRISPR Efficiency of SpCas9 and AsCas12a
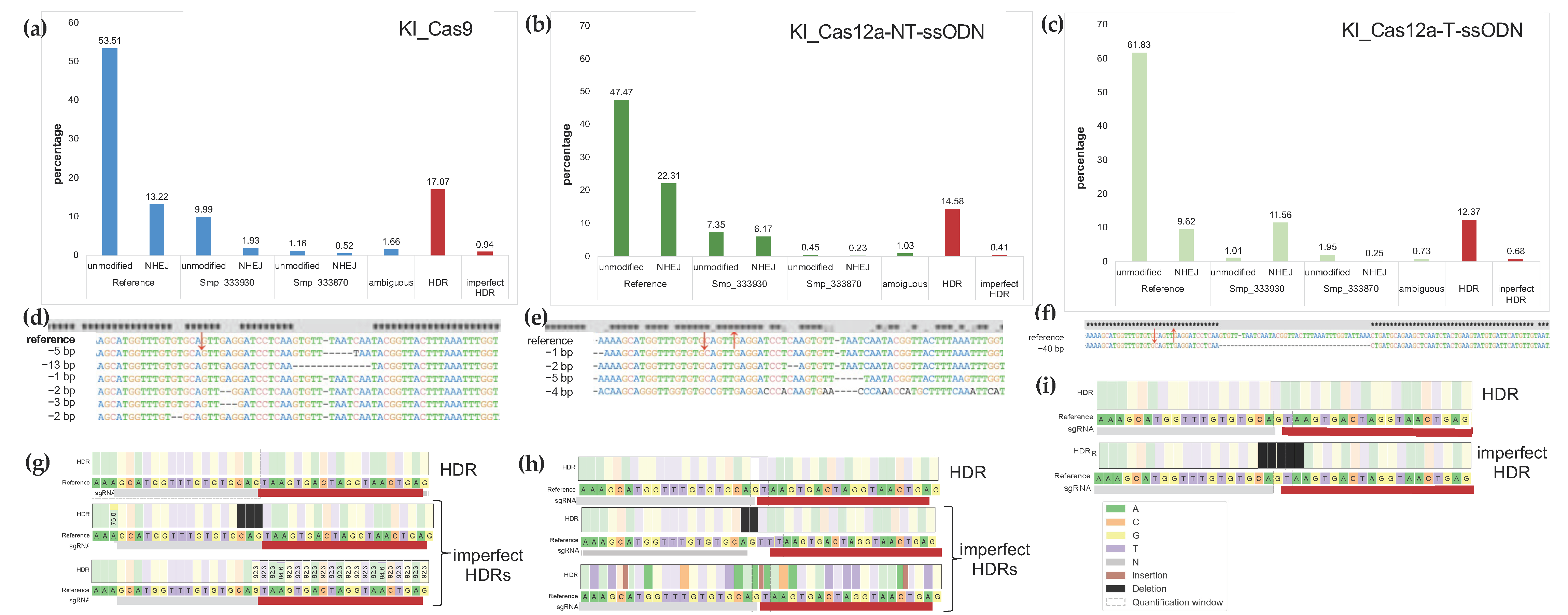
2.5. Estimates of Indels from NHEJ and/or HDR Using NGS Reads from Targeted Amplicons
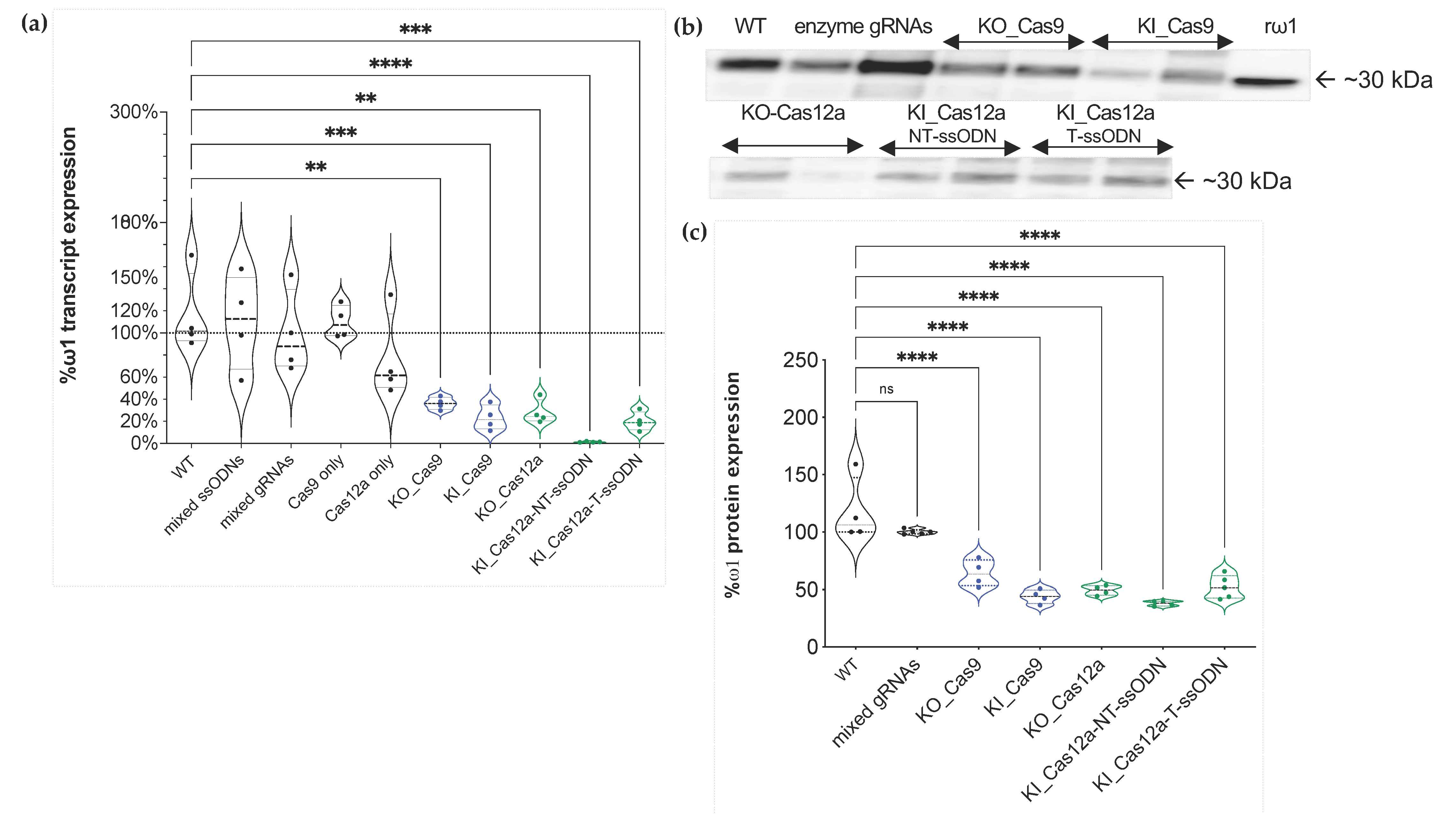
2.6. Transcription of ω1 Interrupted by Programmed Mutation by SpCas9 and AsCas12a
2.7. Protein Levels Ascertained by Western Blotting
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. CRISPR/Cas Target Design and Single-Stranded DNA Donors
4.3. CRISPR Reagents
4.4. Ribonucleoprotein (RNP) Assembly and Delivery by Electroporation
4.5. PCR to Detect a Multiple Stop Codon Containing Transgene
4.6. Real-Time qPCR to Estimate Transgene Copy Numbers
4.7. Tracking of INDELs by Decomposition (TIDE) Analysis
4.8. On-Target Amplicon Next-Generation Sequencing
4.9. Quantitative Real-Time PCR
4.10. Western Blot Analysis
4.11. Single-Stranded DNA Donors as a Potential Substrate for Indiscriminate Trans Activity of Activated AsCas12a
4.12. Statistical Analysis
4.13. Data Submission
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ittiprasert, W.; Mann, V.H.; Karinshak, S.E.; Coghlan, A.; Rinaldi, G.; Sankaranarayanan, G.; Chaidee, A.; Tanno, T.; Kumkhaek, C.; Prangtaworn, P.; et al. Programmed genome editing of the omega-1 ribonuclease of the blood fluke, Schistosoma mansoni. Elife 2019, 8, e41337. [Google Scholar] [CrossRef] [PubMed]
- Arunsan, P.; Ittiprasert, W.; Smout, M.J.; Cochran, C.J.; Mann, V.H.; Chaiyadet, S.; Karinshak, S.E.; Sripa, B.; Young, N.D.; Sotillo, J.; et al. Programmed knockout mutation of liver fluke granulin attenuates virulence of infection-induced hepatobiliary morbidity. Elife 2019, 8, e41463. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.F.; Brindley, P.J.; Berriman, M. Halting harmful helminths. Science 2014, 346, 168–169. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Wolf, Y.I. Evolutionary genomics of defense systems in archaea and bacteria. Annu. Rev. Microbiol. 2017, 71, 233–261. [Google Scholar] [CrossRef]
- Charpentier, E. CRISPR-Cas9: How research on a bacterial RNA-guided mechanism opened new perspectives in biotechnology and biomedicine. EMBO Mol. Med. 2015, 7, 363–365. [Google Scholar] [CrossRef]
- Huang, C.H.; Lee, K.C.; Doudna, J.A. Applications of CRISPR-cas enzymes in cancer therapeutics and detection. Trends Cancer 2018, 4, 499–512. [Google Scholar] [CrossRef]
- Mustafa, M.I.; Makhawi, A.M. SHERLOCK and DETECTR, CRISPR-cas systems as potential rapid diagnostic tools for emerging infectious diseases. J. Clin. Microbiol. 2020, 59, e00745-20. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.; Choi, Y.H.; Jang, Y.; Yu, J.; Goo, J.; Lee, G.; Jeong, Y.K.; Lee, S.H.; Kim, I.S.; Kim, J.S.; et al. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat. Commun. 2018, 9, 2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Malzahn, A.A.; Tang, X.; Lee, K.; Ren, Q.; Sretenovic, S.; Zhang, Y.; Chen, H.; Kang, M.; Bao, Y.; Zheng, X.; et al. Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol. 2019, 17, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, E.; Richter, H.; van der Oost, J.; White, M.F. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-cas adaptive immunity. FEMS Microbiol. Rev. 2015, 39, 428–441. [Google Scholar] [CrossRef]
- Liu, Y.; Han, J.; Chen, Z.; Wu, H.; Dong, H.; Nie, G. Engineering cell signaling using tunable CRISPR-Cpf1-based transcription factors. Nat. Commun. 2017, 8, 2095. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.W.; Nandu, N.; Kachwala, M.J.; Chen, Y.S.; Uyar, T.B.; Yigit, M.V. Probing CRISPR-Cas12a nuclease activity using double-stranded DNA-templated fluorescent substrates. Biochemistry 2020, 59, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. SHERLOCK: Nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef]
- Li, S.-Y.; Cheng, Q.-X.; Wang, J.-M.; Li, X.-Y.; Zhang, Z.-L.; Gao, S.; Cao, R.-B.; Zhao, G.-P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banakar, R.; Schubert, M.; Collingwood, M.; Vakulskas, C.; Eggenberger, A.L.; Wang, K. Comparison of CRISPR-Cas9/Cas12a ribonucleoprotein complexes for genome editing efficiency in the rice phytoene desaturase (OsPDS) gene. Rice 2020, 13, 4. [Google Scholar] [CrossRef]
- Takaki, K.K.; Roca, F.J.; Schramm, G.; Wilbers, R.H.P.; Ittiprasert, W.; Brindley, P.J.; Rinaldi, G.; Berriman, M.; Ramakrishnan, L.; Pagan, A.J. Tumor necrosis factor and Schistosoma mansoni egg antigen omega-1 shape distinct aspects of the early egg-induced granulomatous response. PLoS Negl. Trop. Dis. 2021, 15, e0008814. [Google Scholar] [CrossRef] [PubMed]
- Takaki, K.K.; Rinaldi, G.; Berriman, M.; Pagan, A.J.; Ramakrishnan, L. Schistosoma mansoni eggs modulate the timing of granuloma formation to promote transmission. Cell Host Microbe 2021, 29, 58–67.e5. [Google Scholar] [CrossRef]
- Sankaranarayanan, G.; Berriman, M.; Rinaldi, G. An uneven race: Genome editing for parasitic worms. Nat. Rev. Microbiol. 2021, 19, 621. [Google Scholar] [CrossRef]
- Schwartz, C.; Fallon, P.G. Schistosoma “Eggs-iting” the host: Granuloma formation and egg excretion. Front. Immunol. 2018, 9, 2492. [Google Scholar] [CrossRef] [Green Version]
- Bolt, B.J.; Rodgers, F.H.; Shafie, M.; Kersey, P.J.; Berriman, M.; Howe, K.L. Using wormBase paraSite: An integrated platform for exploring helminth genomic data. Methods Mol. Biol. 2018, 1757, 471–491. [Google Scholar]
- Luhtala, N.; Parker, R. T2 Family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gier, R.A.; Budinich, K.A.; Evitt, N.H.; Cao, Z.; Freilich, E.S.; Chen, Q.; Qi, J.; Lan, Y.; Kohli, R.M.; Shi, J. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 2020, 11, 3455. [Google Scholar] [CrossRef]
- Jacobsen, T.; Ttofali, F.; Liao, C.; Manchalu, S.; Gray, B.N.; Beisel, C.L. Characterization of Cas12a nucleases reveals diverse PAM profiles between closely-related orthologs. Nucleic Acids Res. 2020, 48, 5624–5638. [Google Scholar] [CrossRef] [PubMed]
- Pinello, L.; Canver, M.C.; Hoban, M.D.; Orkin, S.H.; Kohn, D.B.; Bauer, D.E.; Yuan, G.C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016, 34, 695–697. [Google Scholar] [CrossRef] [Green Version]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of validation strategies for CRISPR-Cas9 editing. Sci. Rep. 2018, 8, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.; Geng, Y.; Yao, J.; Fu, C.; Lu, M.; Wang, C.; Du, J. Efficient genome editing in populus using CRISPR/Cas12a. Front. Plant Sci. 2020, 11, 593938. [Google Scholar] [CrossRef]
- Dumeau, C.E.; Monfort, A.; Kissling, L.; Swarts, D.C.; Jinek, M.; Wutz, A. Introducing gene deletions by mouse zygote electroporation of Cas12a/Cpf1. Transgenic Res. 2019, 28, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Kissling, L.; Monfort, A.; Swarts, D.C.; Wutz, A.; Jinek, M. Preparation and electroporation of Cas12a/Cpf1-guide RNA complexes for introducing large gene deletions in mouse embryonic stem cells. Methods Enzymol. 2019, 616, 241–263. [Google Scholar]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J. Cyst. Fibros. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Straume, A.H.; Kjaerner-Semb, E.; Ove Skaftnesmo, K.; Guralp, H.; Kleppe, L.; Wargelius, A.; Edvardsen, R.B. Indel locations are determined by template polarity in highly efficient in vivo CRISPR/Cas9-mediated HDR in Atlantic salmon. Sci. Rep. 2020, 10, 409. [Google Scholar] [CrossRef]
- Steinfelder, S.; Andersen, J.F.; Cannons, J.L.; Feng, C.G.; Joshi, M.; Dwyer, D.; Caspar, P.; Schwartzberg, P.L.; Sher, A.; Jankovic, D. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1). J. Exp. Med. 2009, 206, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Perona-Wright, G.; Smits, H.H.; Hokke, C.H.; van der Ham, A.J.; Fitzsimmons, C.M.; Doenhoff, M.J.; van der Bosch, J.; Mohrs, K.; Haas, H.; et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J. Exp. Med. 2009, 206, 1673–1680. [Google Scholar] [CrossRef] [Green Version]
- Costain, A.H.; MacDonald, A.S.; Smits, H.H. Schistosome egg migration, mechanisms, pathogenesis and host immune responses. Front. Immunol. 2018, 9, 3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- McVeigh, P.; Maule, A.G. Can CRISPR help in the fight against parasitic worms? eLife 2019, 8, e44382. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jinek, M. Mechanistic insights into the cis- and trans-acting DNase activities of Cas12a. Mol. Cell 2019, 73, 589–600.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protasio, A.V.; Tsai, I.J.; Babbage, A.; Nichol, S.; Hunt, M.; Aslett, M.A.; De Silva, N.; Velarde, G.S.; Anderson, T.J.; Clark, R.C.; et al. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012, 6, e1455. [Google Scholar] [CrossRef]
- Swarts, D.C. Making the cut(s): How Cas12a cleaves target and non-target DNA. Biochem. Soc. Trans. 2019, 47, 1499–1510. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jinek, M. Cas9 versus Cas12a/Cpf1, Structure-function comparisons and implications for genome editing. Wiley Interdiscip. Rev. RNA 2018, 9, e1481. [Google Scholar] [CrossRef]
- Canaj, H.; Hussmann, J.A.; Li, H.; Beckman, K.A.; Goodrich, L.; Cho, N.H.; Li, Y.J.; Santos, D.A.; McGeever, A.; Stewart, E.M.; et al. Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in experiments. bioRxiv 2019, 841098. [Google Scholar] [CrossRef] [Green Version]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef] [Green Version]
- Ranawakage, D.C.; Okada, K.; Sugio, K.; Kawaguchi, Y.; Kuninobu-Bonkohara, Y.; Takada, T.; Kamachi, Y. Efficient CRISPR-Cas9-mediated knock-in of composite tags in zebrafish using long ssDNA as a donor. Front. Cell Dev. Biol. 2020, 8, 598634. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Smith, B.M.; Jain, P.K. Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection. Nat. Commun. 2020, 11, 4906. [Google Scholar] [CrossRef]
- Fuchs, R.T.; Curcuru, J.; Mabuchi, M.; Yourik, P.; Robb, G.B. Cas12a trans-cleavage can be modulated in vitro and is active on ssDNA, dsDNA, and RNA. bioRxiv 2019, 600890. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.K.; Kim, K.; Been, K.W.; Baek, G.; Ye, S.; Hur, J.W.; Ryu, S.M.; Lee, Y.S.; Kim, J.S. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 2016, 34, 807–808. [Google Scholar] [CrossRef]
- Tóth, E.; Czene, B.C.; Kulcsár, P.I.; Krausz, S.L.; Tálas, A.; Nyeste, A.; Varga, É.; Huszár, K.; Weinhardt, N.; Ligeti, Z.; et al. Mb- and FnCpf1 nucleases are active in mammalian cells: Activities and PAM preferences of four wild-type Cpf1 nucleases and of their altered PAM specificity variants. Nucleic Acids Res. 2018, 46, 10272–10285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, Q.; Tang, X.; Liu, S.; Malzahn, A.A.; Zhou, J.; Wang, J.; Yin, D.; Pan, C.; Yuan, M.; et al. Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems. Nat. Commun. 2021, 12, 1944. [Google Scholar] [CrossRef]
- Liu, P.; Luk, K.; Shin, M.; Idrizi, F.; Kwok, S.; Roscoe, B.; Mintzer, E.; Suresh, S.; Morrison, K.; Frazão, J.B.; et al. Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res. 2019, 47, 4169–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Meng, Q.; Sun, B.; Zhao, B.; Dang, L.; Zhong, M.; Liu, S.; Xu, H.; Mei, H.; Liu, J.; et al. MeCas12a, a highly sensitive and specific system for COVID-19 detection. Adv. Sci. 2020, 7, 2001300. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.Q.; Soenksen, L.R.; Donghia, N.M.; Angenent-Mari, N.M.; de Puig, H.; Huang, A.; Lee, R.; Slomovic, S.; Galbersanini, T.; Lansberry, G.; et al. Wearable materials with embedded synthetic biology sensors for biomolecule detection. Nat. Biotechnol. 2021, 39, 1366–1374. [Google Scholar] [CrossRef]
- Zamanian, M.; Andersen, E.C. Prospects and challenges of CRISPR/Cas genome editing for the study and control of neglected vector-borne nematode diseases. FEBS J. 2016, 283, 3204–3221. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, J.P.; Day, S.R.; Drew, A.C.; Brindley, P.J. A method for the isolation of schistosome eggs and miracidia free of contaminating host tissues. Parasitology 1997, 115, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Jurberg, A.D.; Goncalves, T.; Costa, T.A.; de Mattos, A.C.; Pascarelli, B.M.; de Manso, P.P.; Ribeiro-Alves, M.; Pelajo-Machado, M.; Peralta, J.M.; Coelho, P.M.; et al. The embryonic development of Schistosoma mansoni eggs: Proposal for a new staging system. Dev. Genes Evol. 2009, 219, 219–234. [Google Scholar] [CrossRef]
- Mann, V.H.; Morales, M.E.; Rinaldi, G.; Brindley, P.J. Culture for genetic manipulation of developmental stages of Schistosoma mansoni. Parasitology 2010, 137, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Mann, V.H.; Suttiprapa, S.; Skinner, D.E.; Brindley, P.J.; Rinaldi, G. Pseudotyped murine leukemia virus for schistosome transgenesis: Approaches, methods and perspectives. Transgenic Res. 2014, 23, 539–556. [Google Scholar] [CrossRef]
- Lu, Z.; Sankaranarayanan, G.; Rawlinson, K.A.; Offord, V.; Brindley, P.J.; Berriman, M.; Rinaldi, G. The transcriptome of Schistosoma mansoni developing eggs reveals key mediators in pathogenesis and life cycle propagation. Front. Trop. Dis. 2021, 2, 713123. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirvani, R.; Barshan-Tashnizi, M.; Shahali, M. An investigation into gene copy number determination in transgenic yeast; The importance of selecting a reliable real-time PCR standard. Biologicals 2020, 65, 10–17. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Shin, S.G.; Hwang, S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 2006, 123, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.S.; Burris, J.; Stewart, N.R.; Mentewab, A.; Stewart, C.N., Jr. Statistical tools for transgene copy number estimation based on real-time PCR. BMC Bioinform. 2007, 8 (Suppl. 7), S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshalkina, D.A.; Glushchenko, A.S.; Kysil, E.V.; Mizgirev, I.V.; Frolov, A. SpCas9- and LbCas12a-mediated DNA editing produce different gene knockout outcomes in zebrafish embryos. Genes 2020, 11, 740. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; van Steensel, B. Rapid Quantitative Evaluation of CRISPR Genome Editing by TIDE and TIDER. Methods Mol. Biol. 2019, 1961, 29–44. [Google Scholar] [PubMed] [Green Version]
- Germini, D.; Tsfasman, T.; Zakharova, V.V.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A Comparison of Techniques to Evaluate the Effectiveness of Genome Editing. Trends Biotechnol. 2018, 36, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Bradford assay for determining protein concentration. Cold Spring Harb. Protoc. 2020, 4, 102269. [Google Scholar] [CrossRef] [PubMed]
- Wilbers, R.H.; Westerhof, L.B.; van Noort, K.; Obieglo, K.; Driessen, N.N.; Everts, B.; Gringhuis, S.I.; Schramm, G.; Goverse, A.; Smant, G.; et al. Production and glyco-engineering of immunomodulatory helminth glycoproteins in plants. Sci. Rep. 2017, 7, 45910. [Google Scholar] [CrossRef] [Green Version]
- Bandrowski, A.; Brush, M.; Grethe, J.S.; Haendel, M.A.; Kennedy, D.N.; Hill, S.; Hof, P.R.; Martone, M.E.; Pols, M.; Tan, S.C.; et al. The Resource Identification Initiative: A cultural shift in publishing. Brain Behav. 2016, 6, e00417. [Google Scholar] [CrossRef]
- Li, B.; Yan, J.; Zhang, Y.; Li, W.; Zeng, C.; Zhao, W.; Hou, X.; Zhang, C.; Dong, Y. CRISPR-Cas12a possesses unconventional DNase activity that can be inactivated by synthetic oligonucleotides. Mol. Ther. Nucleic. Acids 2020, 19, 1043–1052. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ittiprasert, W.; Chatupheeraphat, C.; Mann, V.H.; Li, W.; Miller, A.; Ogunbayo, T.; Tran, K.; Alrefaei, Y.N.; Mentink-Kane, M.; Brindley, P.J. RNA-Guided AsCas12a- and SpCas9-Catalyzed Knockout and Homology Directed Repair of the Omega-1 Locus of the Human Blood Fluke, Schistosoma mansoni. Int. J. Mol. Sci. 2022, 23, 631. https://doi.org/10.3390/ijms23020631
Ittiprasert W, Chatupheeraphat C, Mann VH, Li W, Miller A, Ogunbayo T, Tran K, Alrefaei YN, Mentink-Kane M, Brindley PJ. RNA-Guided AsCas12a- and SpCas9-Catalyzed Knockout and Homology Directed Repair of the Omega-1 Locus of the Human Blood Fluke, Schistosoma mansoni. International Journal of Molecular Sciences. 2022; 23(2):631. https://doi.org/10.3390/ijms23020631
Chicago/Turabian StyleIttiprasert, Wannaporn, Chawalit Chatupheeraphat, Victoria H. Mann, Wenhui Li, André Miller, Taiwo Ogunbayo, Kenny Tran, Yousef N. Alrefaei, Margaret Mentink-Kane, and Paul J. Brindley. 2022. "RNA-Guided AsCas12a- and SpCas9-Catalyzed Knockout and Homology Directed Repair of the Omega-1 Locus of the Human Blood Fluke, Schistosoma mansoni" International Journal of Molecular Sciences 23, no. 2: 631. https://doi.org/10.3390/ijms23020631