
Primary Founder Mutations in the PRKDC Gene Increase Tumor Mutation Load in Colorectal Cancer

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
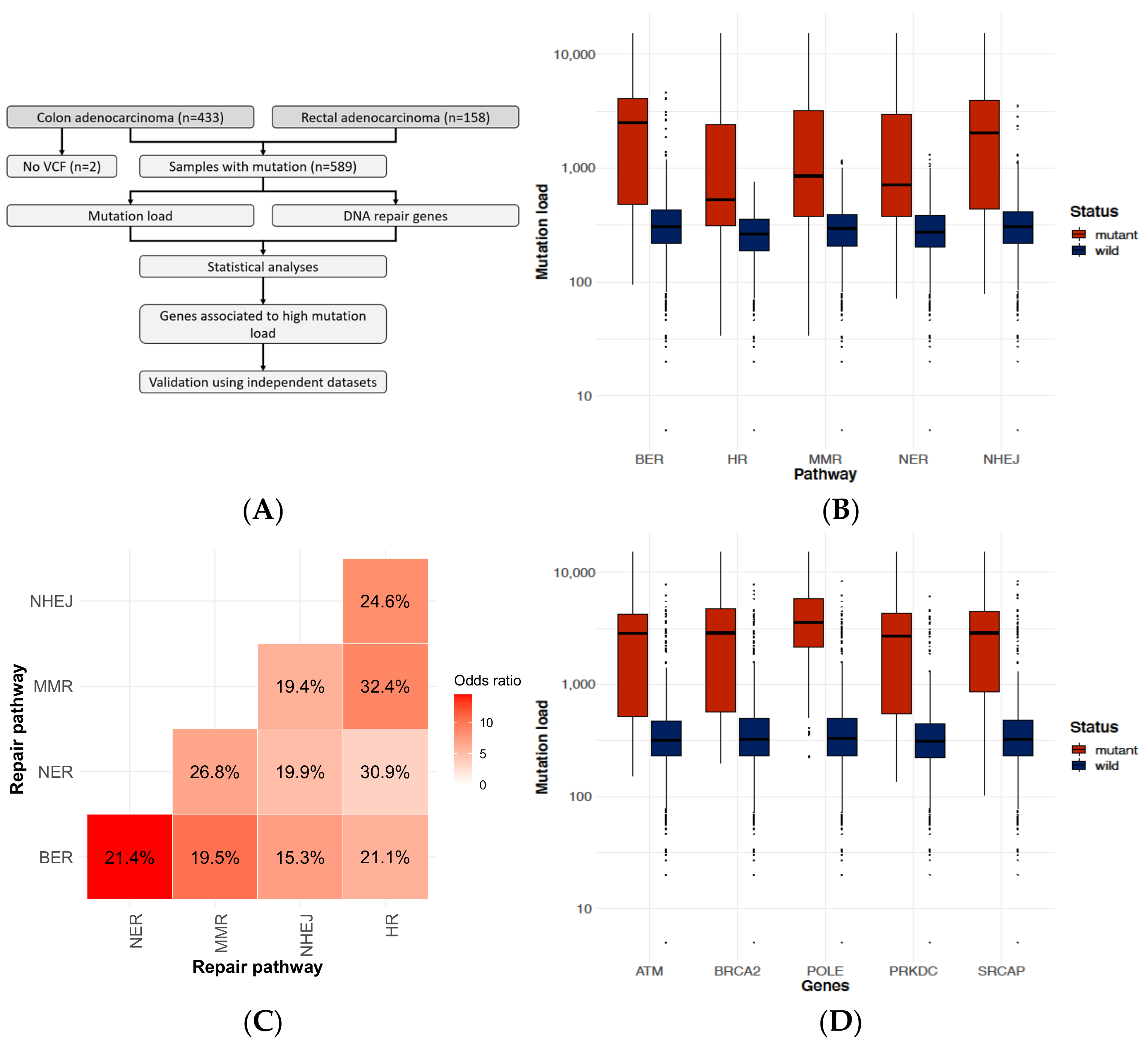
2.1. DNA-Repair Gene Defects Cause an Increased Accumulation Rate of Mutations
2.2. Identifying Genes Where the Mutation Status Is Associated with High Mutation Burden
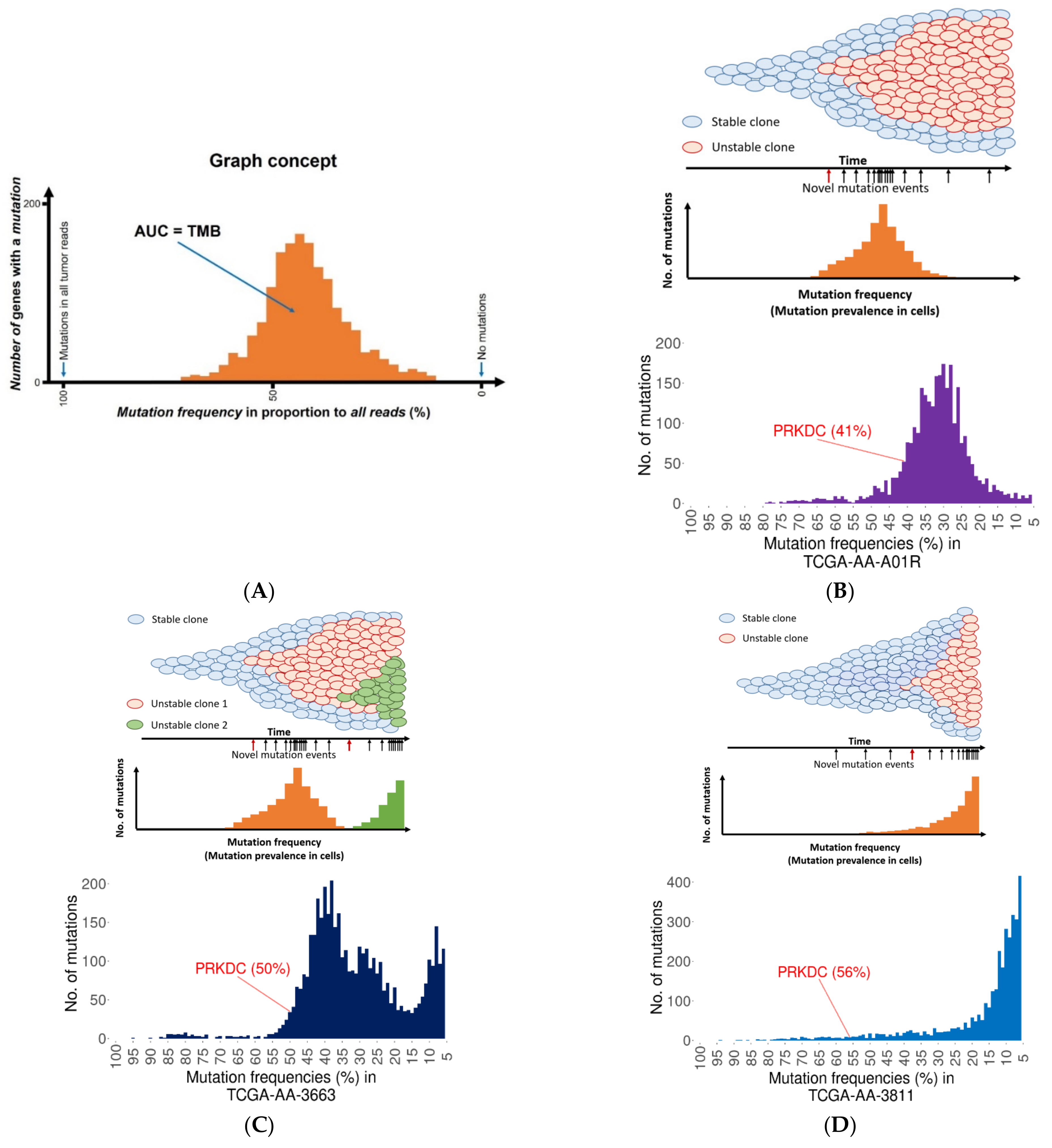
2.3. Founder Mutation and Increased Mutational Load
2.4. PRKDC Acting as a Mutator Phenotype
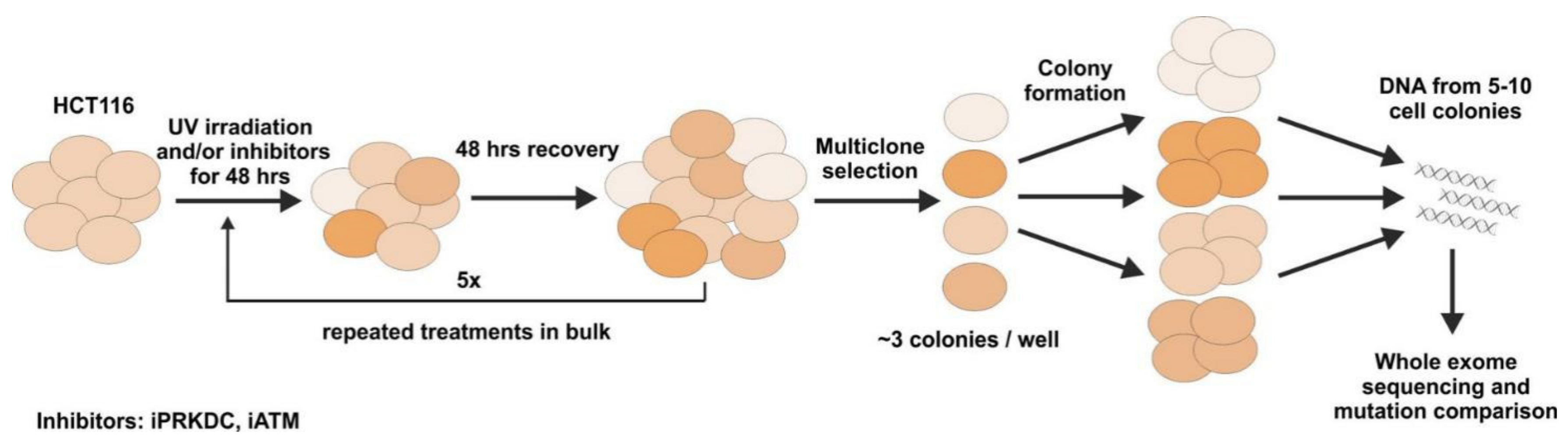
2.5. Mutagenesis Experiment
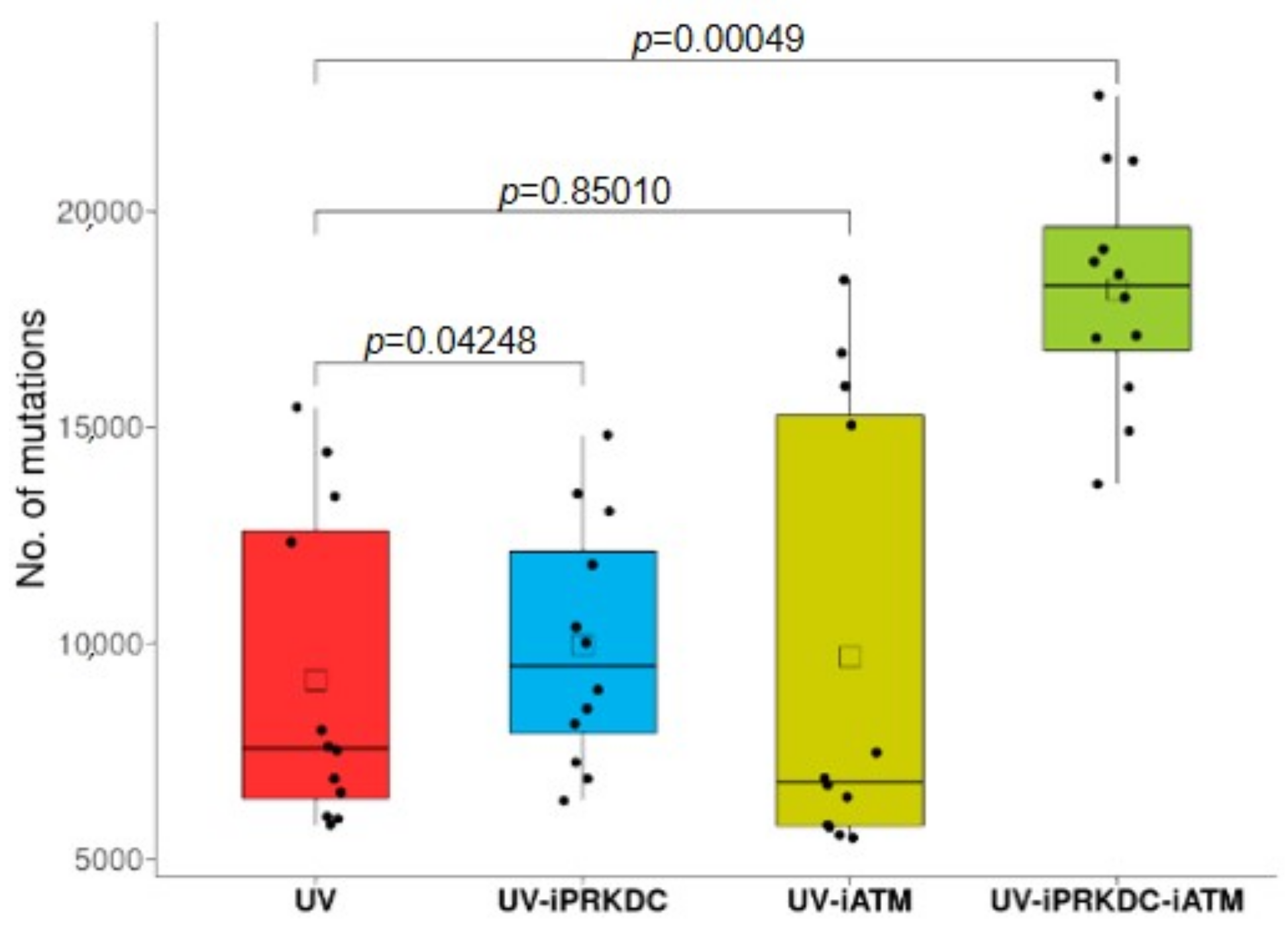
2.6. Effect of PRKDC and ATM1 Inhibition on Mutation Rate in UV-Treated Cell Lines
2.7. Expression of DNA-Repair Genes and Survival Differences
3. Discussion
4. Materials and Methods
4.1. Repair Gene Database Setup
4.2. Mutation Database Setup
4.3. Calculating Correlation between Patient Mutation Burden and Repair Gene Statuses
4.4. Identifying DNA-Repair Associated Founder Mutations
4.5. Cell Culture Setup
4.6. Mutagenesis Experiments
4.7. Analysis of Whole Exome Sequencing Data
4.8. Construction of Gene Expression Database
4.9. Analysis of Clinical Samples
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by P53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Geske, F.J.; Nelson, A.C.; Lieberman, R.; Strange, R.; Sun, T.; Gerschenson, L.E. DNA Repair Is Activated in Early Stages of P53-Induced Apoptosis. Cell Death Differ. 2000, 7, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved Modes of Recruitment of ATM, ATR and DNA-PKcs to Sites of DNA Damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Meek, K.; Dang, V.; Lees-Miller, S.P. Chapter 2 DNA-PK: The Means to Justify the Ends? In Advances in Immunology; Academic Press: New York, NY, USA, 2008; Volume 99, pp. 33–58. [Google Scholar]
- Björkman, A.; Du, L.; Felgentreff, K.; Rosner, C.; Kamdar, R.P.; Kokaraki, G.; Matsumoto, Y.; Davies, E.G.; Burg, M.; van der Notarangelo, L.D.; et al. DNA-PKcs Is Involved in Ig Class Switch Recombination in Human B Cells. J. Immunol. 2015, 195, 5608–5615. [Google Scholar] [CrossRef] [Green Version]
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W. Functional Redundancy between the XLF and DNA-PKcs DNA Repair Factors in V(D)J Recombination and Nonhomologous DNA End Joining. Proc. Natl. Acad. Sci. USA 2013, 110, 2234–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors Determining DNA Double-Strand Break Repair Pathway Choice in G2 Phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive Competition Between Homologous Recombination and Non-Homologous End Joining11NIH Grant CA77693 to J.A.N. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar]
- Uematsu, N.; Weterings, E.; Yano, K.; Morotomi-Yano, K.; Jakob, B.; Taucher-Scholz, G.; Mari, P.-O.; van Gent, D.C.; Chen, B.P.C.; Chen, D.J. Autophosphorylation of DNA-PKCS Regulates Its Dynamics at DNA Double-Strand Breaks. J. Cell Biol. 2007, 177, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Yu, Y.; Gupta, S.; Cho, Y.-M.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of DNA-Dependent Protein Kinase Regulates DNA End Processing and May Also Alter Double-Strand Break Repair Pathway Choice. Mol. Cell. Biol. 2005, 25, 10842–10852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oorschot, B.; Granata, G.; Di Franco, S.; Ten Cate, R.; Rodermond, H.M.; Todaro, M.; Medema, J.P.; Franken, N.A.P. Targeting DNA Double Strand Break Repair with Hyperthermia and DNA-PKcs Inhibition to Enhance the Effect of Radiation Treatment. Oncotarget 2016, 7, 65504–65513. [Google Scholar] [CrossRef] [Green Version]
- Pinel, B.; Duchesne, M.; Godet, J.; Milin, S.; Berger, A.; Wager, M.; Karayan-Tapon, L. Mesenchymal Subtype of Glioblastomas with High DNA-PKcs Expression Is Associated with Better Response to Radiotherapy and Temozolomide. J. Neurooncol. 2017, 132, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Tan, K.T.; Cheng, J.H.; Fang, W.-L.; Yeh, Y.C.; Yeh, C.-N.; Chang, Y.C.; Chen, S.-J.; Hsiao, M.; Chao, Y. PRKDC: A New Candidate for Checkpoint Blockade Immunotherapy? J. Clin. Oncol. 2017, 35, 3022. [Google Scholar] [CrossRef]
- Tan, K.T.; Yeh, C.-N.; Chang, Y.-C.; Cheng, J.-H.; Fang, W.-L.; Yeh, Y.-C.; Wang, Y.-C.; Hsu, D.S.-S.; Wu, C.-E.; Lai, J.-I.; et al. PRKDC: New Biomarker and Drug Target for Checkpoint Blockade Immunotherapy. J. Immunother. Cancer 2020, 8, e000485. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ma, Y.; Ma, L.; Guan, Y.; Ma, L.; Yang, D. MicroRNA-488-3p Sensitizes Malignant Melanoma Cells to Cisplatin by Targeting PRKDC. Cell Biol. Int. 2017, 41, 622–629. [Google Scholar] [CrossRef]
- Mamo, T.; Mladek, A.C.; Shogren, K.L.; Gustafson, C.; Gupta, S.K.; Riester, S.M.; Maran, A.; Galindo, M.; van Wijnen, A.J.; Sarkaria, J.N.; et al. Inhibiting DNA-PKCS Radiosensitizes Human Osteosarcoma Cells. Biochem. Biophys. Res. Commun. 2017, 486, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Gurley, K.E.; Ashley, A.K.; Moser, R.D.; Kemp, C.J. Synergy between Prkdc and Trp53 Regulates Stem Cell Proliferation and GI-ARS after Irradiation. Cell Death Differ. 2017, 24, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Yang, L.; Dong, C.; Ma, B.; Shan, M.; Ma, B. PRKDC Regulates Chemosensitivity and Is a Potential Prognostic and Predictive Marker of Response to Adjuvant Chemotherapy in Breast Cancer Patients. Oncol. Rep. 2017, 37, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.; Arora, A.; Agarwal, D.; Moseley, P.; Perry, C.; Thompson, N.; Green, A.R.; Rakha, E.; Chan, S.; Ball, G.; et al. Adverse Prognostic and Predictive Significance of Low DNA-Dependent Protein Kinase Catalytic Subunit (DNA-PKcs) Expression in Early-Stage Breast Cancers. Breast Cancer Res. Treat. 2014, 146, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.-H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Finzel, A.; Grybowski, A.; Strasen, J.; Cristiano, E.; Loewer, A. Hyperactivation of ATM upon DNA-PKcs Inhibition Modulates P53 Dynamics and Cell Fate in Response to DNA Damage. Mol. Biol. Cell 2016, 27, 2360–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Sibanda, B.L.; Chirgadze, D.Y.; Ascher, D.B.; Blundell, T.L. DNA-PKcs Structure Suggests an Allosteric Mechanism Modulating DNA Double-Strand Break Repair. Science 2017, 355, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Albarakati, N.; Abdel-Fatah, T.M.A.; Doherty, R.; Russell, R.; Agarwal, D.; Moseley, P.; Perry, C.; Arora, A.; Alsubhi, N.; Seedhouse, C.; et al. Targeting BRCA1-BER Deficient Breast Cancer by ATM or DNA-PKcs Blockade Either Alone or in Combination with Cisplatin for Personalized Therapy. Mol. Oncol. 2015, 9, 204–217. [Google Scholar] [CrossRef]
- Gupta, D.; Heinen, C.D. The Mismatch Repair-Dependent DNA Damage Response: Mechanisms and Implications. DNA Repair 2019, 78, 60–69. [Google Scholar] [CrossRef]
- Germano, G.; Amirouchene-Angelozzi, N.; Rospo, G.; Bardelli, A. The Clinical Impact of the Genomic Landscape of Mismatch Repair-Deficient Cancers. Cancer Discov. 2018, 8, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch Repair Deficiency Commonly Precedes Adenoma Formation in Lynch Syndrome-Associated Colorectal Tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Langenbucher, A.; Bowen, D.; Sakhtemani, R.; Bournique, E.; Wise, J.F.; Zou, L.; Bhagwat, A.S.; Buisson, R.; Lawrence, M.S. An Extended APOBEC3A Mutation Signature in Cancer. Nat. Commun. 2021, 12, 1602. [Google Scholar] [CrossRef]
- Shi, M.-J.; Meng, X.-Y.; Fontugne, J.; Chen, C.-L.; Radvanyi, F.; Bernard-Pierrot, I. Identification of New Driver and Passenger Mutations within APOBEC-Induced Hotspot Mutations in Bladder Cancer. Genome Med. 2020, 12, 85. [Google Scholar] [CrossRef]
- Buisson, R.; Langenbucher, A.; Bowen, D.; Kwan, E.E.; Benes, C.H.; Zou, L.; Lawrence, M.S. Passenger Hotspot Mutations in Cancer Driven by APOBEC3A and Mesoscale Genomic Features. Science 2019, 364, eaaw2872. [Google Scholar] [CrossRef] [PubMed]
- Cannataro, V.L.; Gaffney, S.G.; Sasaki, T.; Issaeva, N.; Grewal, N.K.S.; Grandis, J.R.; Yarbrough, W.G.; Burtness, B.; Anderson, K.S.; Townsend, J.P. APOBEC-Induced Mutations and Their Cancer Effect Size in Head and Neck Squamous Cell Carcinoma. Oncogene 2019, 38, 3475–3487. [Google Scholar] [CrossRef] [Green Version]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Zhao, J.; Xia, H.; Xie, J.; Guo, Y.; Zhong, L.; Li, M.; Yang, Q.; Peng, C.; et al. DNA-PK Deficiency Potentiates CGAS-Mediated Antiviral Innate Immunity. Nat. Commun. 2020, 11, 6182. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766.e9. [Google Scholar] [CrossRef]
- Affandi, T.; Ohm, A.M.; Gaillard, D.; Haas, A.; Reyland, M.E. Tyrosine Kinase Inhibitors Protect the Salivary Gland from Radiation Damage by Increasing DNA Double Strand Break Repair. J. Biol. Chem. 2021, 296, 100401. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, S.; Zhang, W.; Li, Z.; Xu, J.; Li, D.; Wang, Y.; Zhan, Q. A Comprehensive Analysis of Alterations in DNA Damage Repair Pathways Reveals a Potential Way to Enhance the Radio-Sensitivity of Esophageal Squamous Cell Cancer. Front. Oncol. 2020, 10, 575711. [Google Scholar] [CrossRef]
- Wang, C.; Tang, H.; Geng, A.; Dai, B.; Zhang, H.; Sun, X.; Chen, Y.; Qiao, Z.; Zhu, H.; Yang, J.; et al. Rational Combination Therapy for Hepatocellular Carcinoma with PARP1 and DNA-PK Inhibitors. Proc. Natl. Acad. Sci. USA 2020, 117, 26356–26365. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A Platform for Interactive Large-Scale Genome Analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Molnar, B.; Lage, H.; Szallasi, Z.; Eklund, A.C. Evaluation of Microarray Preprocessing Algorithms Based on Concordance with RT-PCR in Clinical Samples. PLoS ONE 2009, 4, e5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Birkbak, N.J.; Gyorffy, B.; Szallasi, Z.; Eklund, A.C. Jetset: Selecting the Optimal Microarray Probe Set to Represent a Gene. BMC Bioinform. 2011, 12, 474. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Gene | p-Value | Mutation Burden (Mutant Samples) | Mutation Burden (Wild Samples) | Fold Increase | Samples with Mutation (%) | Total Samples |
|---|---|---|---|---|---|---|---|
| TCGA COAD (mutect2) | PRKDC | 9.5 × 10−28 | 2853 | 329 | 8.7 | 22.0 | 431 |
| ATM | 3.7 × 10−24 | 2926 | 333 | 8.8 | 20.0 | 431 | |
| POLE | 1.3 × 10−22 | 4002 | 336 | 11.9 | 13.9 | 431 | |
| BRCA2 | 1.2 × 10−20 | 2951 | 335 | 8.8 | 15.1 | 431 | |
| POLD1 | 5.1 × 10−18 | 3538 | 348 | 10.2 | 10.2 | 431 | |
| DFCI | POLE | 2.2 × 10−14 | 919 | 123 | 7.5 | 7.3 | 619 |
| BRCA2 | 1.2 × 10−13 | 948 | 123 | 7.7 | 6.3 | 619 | |
| ATM | 1.4 × 10−12 | 819 | 123 | 6.7 | 7.4 | 619 | |
| PRKDC | 1.3 × 10−9 | 765 | 124 | 6.2 | 6.8 | 619 | |
| MLH3 | 1.1 × 10−8 | 1122 | 126 | 8.9 | 3.2 | 619 | |
| GenenTech | ATM | 4.4 × 10−4 | 909 | 82 | 11.1 | 18.1 | 72 |
| RAD50 | 1.2 × 10−3 | 2393 | 86 | 27.8 | 5.6 | 72 | |
| BRCA2 | 1.5 × 10−3 | 1218 | 84 | 14.5 | 6.9 | 72 | |
| PRKDC | 2.2 × 10−3 | 1671 | 86 | 19.4 | 5.6 | 72 | |
| LIG1 | 3.2 × 10−3 | 1195 | 84 | 14.2 | 6.9 | 72 |
| Gene | HR | CI | p-Value | Expression Fold Change (Tumor vs. Normal) |
|---|---|---|---|---|
| PRKDC | 0.72 | 0.58–0.9 | 4.40 × 10−3 | 6.44 |
| BRCA2 | 0.73 | 0.58–0.91 | 5.60 × 10−3 | 4.30 |
| RAD50 | 0.81 | 0.64–1.02 | 6.90 × 10−2 | 2.74 |
| ATM | 1.18 | 0.93–1.49 | 1.68 × 10−1 | 1.48 |
| LIG1 | 1.17 | 0.94–1.46 | 1.70 × 10−1 | 1.40 |
| MLH3 | 1.48 | 1.13–1.95 | 4.70 × 10−3 | 1.38 |
| POLD1 | 0.67 | 0.53–0.85 | 8.0 × 10−4 | 1.09 |
| POLE | 0.61 | 0.47–0.77 | 5.30 × 10−5 | 0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pálinkás, H.L.; Pongor, L.; Balajti, M.; Nagy, Á.; Nagy, K.; Békési, A.; Bianchini, G.; Vértessy, B.G.; Győrffy, B. Primary Founder Mutations in the PRKDC Gene Increase Tumor Mutation Load in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 633. https://doi.org/10.3390/ijms23020633
Pálinkás HL, Pongor L, Balajti M, Nagy Á, Nagy K, Békési A, Bianchini G, Vértessy BG, Győrffy B. Primary Founder Mutations in the PRKDC Gene Increase Tumor Mutation Load in Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(2):633. https://doi.org/10.3390/ijms23020633
Chicago/Turabian StylePálinkás, Hajnalka Laura, Lőrinc Pongor, Máté Balajti, Ádám Nagy, Kinga Nagy, Angéla Békési, Giampaolo Bianchini, Beáta G. Vértessy, and Balázs Győrffy. 2022. "Primary Founder Mutations in the PRKDC Gene Increase Tumor Mutation Load in Colorectal Cancer" International Journal of Molecular Sciences 23, no. 2: 633. https://doi.org/10.3390/ijms23020633
APA StylePálinkás, H. L., Pongor, L., Balajti, M., Nagy, Á., Nagy, K., Békési, A., Bianchini, G., Vértessy, B. G., & Győrffy, B. (2022). Primary Founder Mutations in the PRKDC Gene Increase Tumor Mutation Load in Colorectal Cancer. International Journal of Molecular Sciences, 23(2), 633. https://doi.org/10.3390/ijms23020633