
Bisphenol S Impairs Invasion and Proliferation of Extravillous Trophoblasts Cells by Interfering with Epidermal Growth Factor Receptor Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
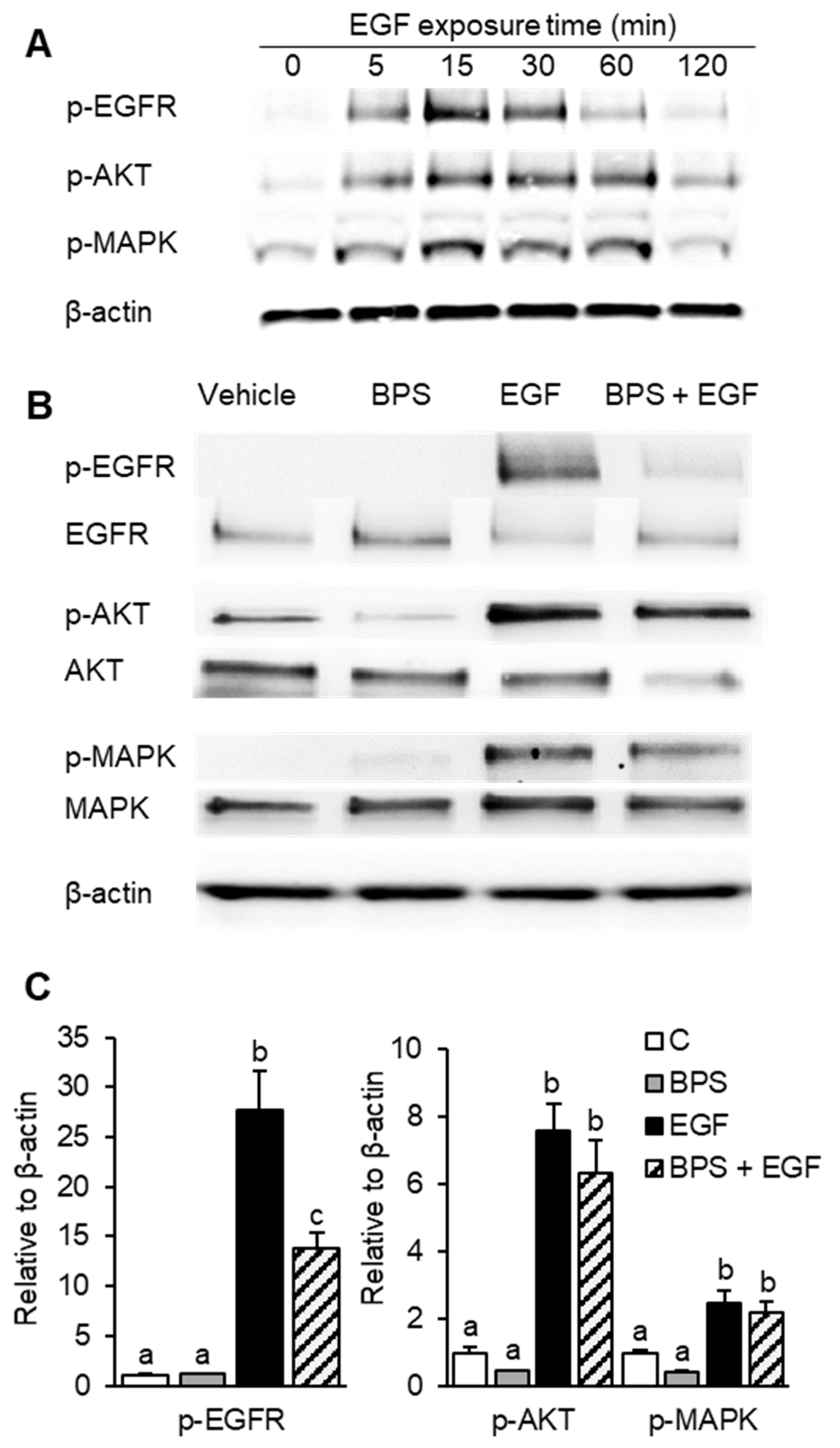
2.1. BPS Inhibits EGFR Phosphorylation
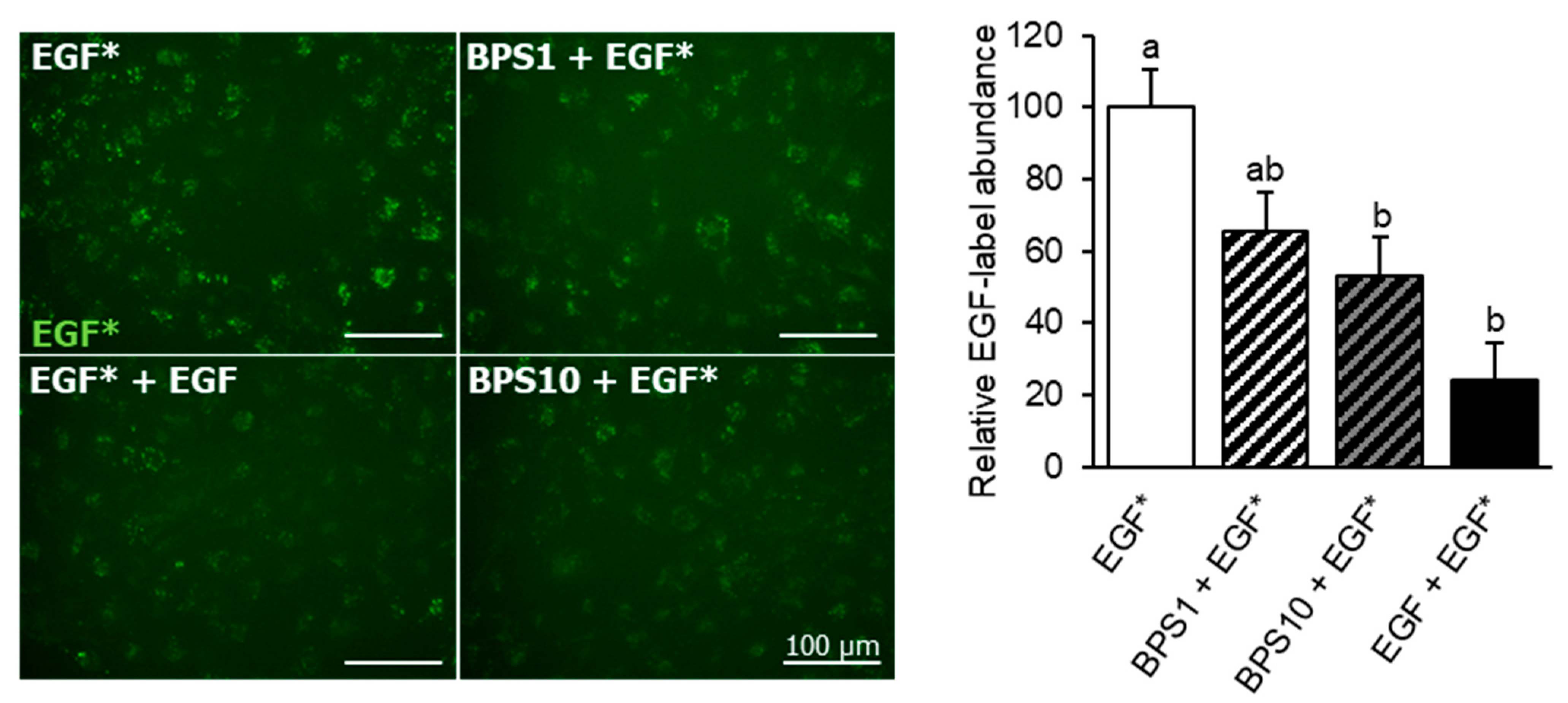
2.2. BPS Reduces EGF Internalization
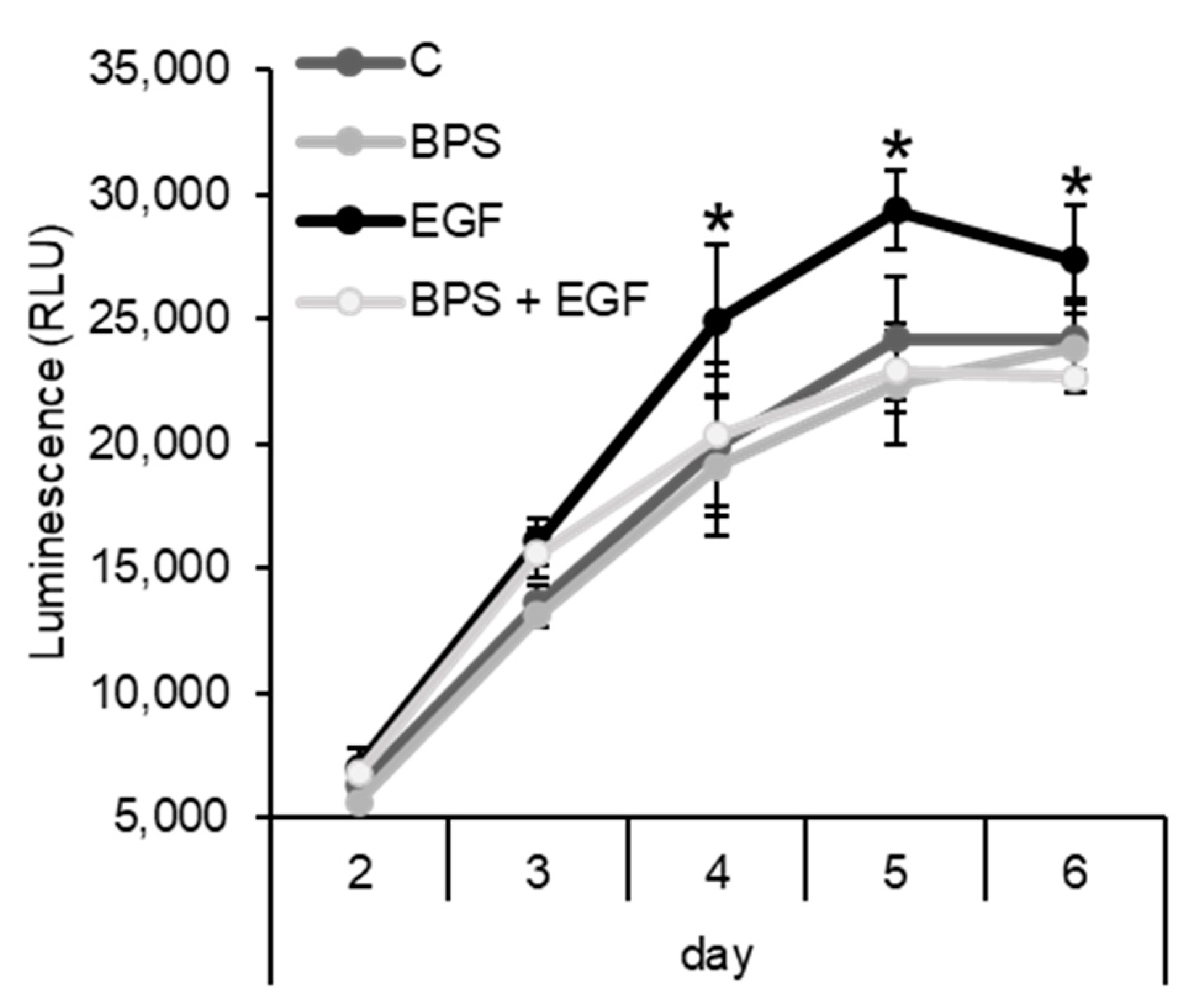
2.3. BPS Impairs EGF-Mediated Cell Proliferation
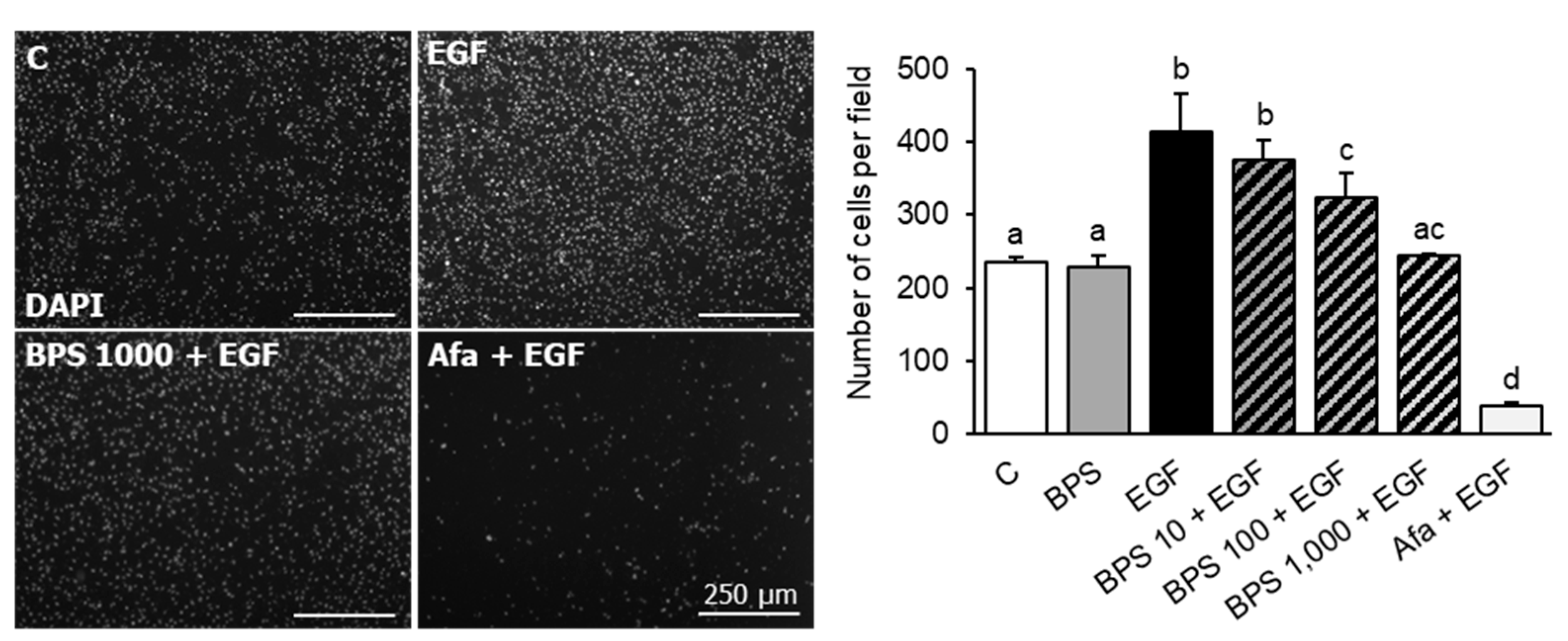
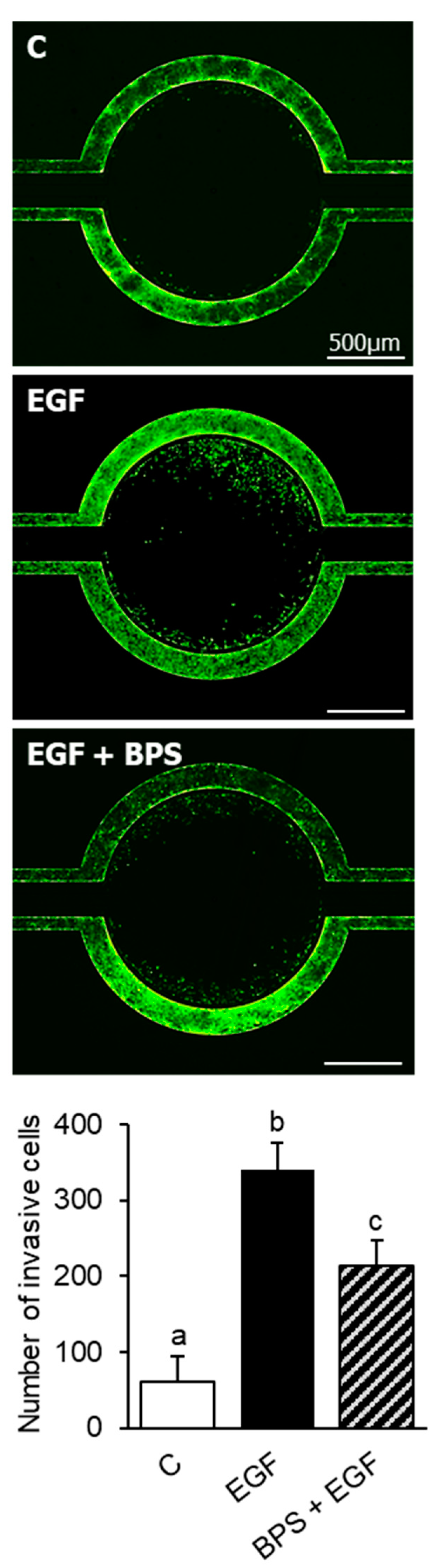
2.4. BPS Impairs EGF-Mediated Cell Invasion
2.5. Tube Forming
3. Discussion
4. Materials and Methods
4.1. Exposure Chemicals
4.2. HTR-8/SVneo Cell Culture
4.3. Cell Proliferation Assay
4.4. Western Blotting
4.5. EGF Endocytosis Assay
4.6. Transwell Cell Invasion
4.7. 3D Microfluidic Chip Cell Invasion
4.8. Endovascular Differentiation Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- LaRocca, J.; Binder, A.M.; McElrath, T.F.; Michels, K.B. First-Trimester Urine Concentrations of Phthalate Metabolites and Phenols and Placenta miRNA Expression in a Cohort of U.S. Women. Environ. Health Perspect. 2016, 124, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, J.; Ticiani, E.; Veiga-Lopez, A. Placenta Disrupted: Endocrine Disrupting Chemicals and Pregnancy. Trends Endocrinol. Metab. 2020, 31, 508–524. [Google Scholar] [CrossRef]
- Birnbaum, L.S. State of the science of endocrine disruptors. Environ. Health Perspect. 2013, 121, A107. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- US-EPA/United States Environmental Protection Agency. Assessing and Managing Chemicals under TSCA—Risk Management for Bisphenol A (BPA). 2016. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/risk-management-bisphenol-bpa (accessed on 4 November 2021).
- Liao, C.; Liu, F.; Alomirah, H.; Loi, V.D.; Mohd, M.A.; Moon, H.B.; Nakata, H.; Kannan, K. Bisphenol S in urine from the United States and seven Asian countries: Occurrence and human exposures. Environ. Sci. Technol. 2012, 46, 6860–6866. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulos, A.G.; Xue, J.; De Carvalho, B.P.; Iyer, A.; Abualnaja, K.O.; Yaghmoor, S.S.; Kumosani, T.A.; Kannan, K. Urinary biomarkers of exposure to 57 xenobiotics and its association with oxidative stress in a population in Jeddah, Saudi Arabia. Environ. Res. 2016, 150, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Liu, F.; Moon, H.B.; Yamashita, N.; Yun, S.; Kannan, K. Bisphenol analogues in sediments from industrialized areas in the United States, Japan, and Korea: Spatial and temporal distributions. Environ. Sci. Technol. 2012, 46, 11558–11565. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wong, L.Y.; Kramer, J.; Zhou, X.; Jia, T.; Calafat, A.M. Urinary Concentrations of Bisphenol A and Three Other Bisphenols in Convenience Samples of U.S. Adults during 2000-2014. Environ. Sci. Technol. 2015, 49, 11834–11839. [Google Scholar] [CrossRef]
- Hehn, R.S. NHANES Data Support Link between Handling of Thermal Paper Receipts and Increased Urinary Bisphenol A Excretion. Environ. Sci. Technol. 2016, 50, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Kannan, K. Concentrations and profiles of bisphenol A and other bisphenol analogues in foodstuffs from the United States and their implications for human exposure. J. Agric. Food Chem. 2013, 61, 4655–4662. [Google Scholar] [CrossRef]
- Liao, C.; Liu, F.; Guo, Y.; Moon, H.B.; Nakata, H.; Wu, Q.; Kannan, K. Occurrence of eight bisphenol analogues in indoor dust from the United States and several Asian countries: Implications for human exposure. Environ. Sci. Technol. 2012, 46, 9138–9145. [Google Scholar] [CrossRef]
- Yu, X.; Xue, J.; Yao, H.; Wu, Q.; Venkatesan, A.K.; Halden, R.U.; Kannan, K. Occurrence and estrogenic potency of eight bisphenol analogs in sewage sludge from the U.S. EPA targeted national sewage sludge survey. J. Hazard. Mater. 2015, 299, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, E.; Yamashita, N.; Taniyasu, S.; Lam, J.; Lam, P.K.; Moon, H.B.; Jeong, Y.; Kannan, P.; Achyuthan, H.; Munuswamy, N.; et al. Bisphenol A and other bisphenol analogues including BPS and BPF in surface water samples from Japan, China, Korea and India. Ecotoxicol. Environ. Saf. 2015, 122, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Mersha, M.D.; Patel, B.M.; Patel, D.; Richardson, B.N.; Dhillon, H.S. Effects of BPA and BPS exposure limited to early embryogenesis persist to impair non-associative learning in adults. Behav. Brain Funct. 2015, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speidel, J.T.; Xu, M.; Abdel-Rahman, S.Z. Bisphenol A (BPA) and bisphenol S (BPS) alter the promoter activity of the ABCB1 gene encoding P-glycoprotein in the human placenta in a haplotype-dependent manner. Toxicol. Appl. Pharm. 2018, 359, 47–54. [Google Scholar] [CrossRef]
- Wu, L.H.; Zhang, X.M.; Wang, F.; Gao, C.J.; Chen, D.; Palumbo, J.R.; Guo, Y.; Zeng, E.Y. Occurrence of bisphenol S in the environment and implications for human exposure: A short review. Sci. Total Environ. 2018, 615, 87–98. [Google Scholar] [CrossRef]
- Hill, C.E.; Sapouckey, S.A.; Suvorov, A.; Vandenberg, L.N. Developmental exposures to bisphenol S, a BPA replacement, alter estrogen-responsiveness of the female reproductive tract: A pilot study. Cogent Med. 2017, 4, 1317690. [Google Scholar] [CrossRef]
- Gingrich, J.; Pu, Y.; Ehrhardt, R.; Karthikraj, R.; Kannan, K.; Veiga-Lopez, A. Toxicokinetics of bisphenol A, bisphenol S, and bisphenol F in a pregnancy sheep model. Chemosphere 2019, 220, 185–194. [Google Scholar] [CrossRef]
- Benincasa, L.; Mandala, M.; Paulesu, L.; Barberio, L.; Ietta, F. Prenatal Nutrition Containing Bisphenol A Affects Placenta Glucose Transfer: Evidence in Rats and Human Trophoblast. Nutrients 2020, 12, 1375. [Google Scholar] [CrossRef]
- Narciso, L.; Ietta, F.; Romagnoli, R.; Paulesu, L.; Mantovani, A.; Tait, S. Effects of Bisphenol A on endogenous retroviral envelopes expression and trophoblast fusion in BeWo cells. Reprod. Toxicol. 2019, 89, 35–44. [Google Scholar] [CrossRef]
- Mao, J.; Jain, A.; Denslow, N.D.; Nouri, M.Z.; Chen, S.; Wang, T.; Zhu, N.; Koh, J.; Sarma, S.J.; Sumner, B.W.; et al. Bisphenol A and bisphenol S disruptions of the mouse placenta and potential effects on the placenta-brain axis. Proc. Natl. Acad. Sci. USA 2020, 117, 4642–4652. [Google Scholar] [CrossRef]
- Gingrich, J.; Pu, Y.; Roberts, J.; Karthikraj, R.; Kannan, K.; Ehrhardt, R.; Veiga-Lopez, A. Gestational bisphenol S impairs placental endocrine function and the fusogenic trophoblast signaling pathway. Arch. Toxicol. 2018, 92, 1861–1876. [Google Scholar] [CrossRef]
- Ticiani, E.; Gingrich, J.; Pu, Y.; Vettathu, M.; Davis, J.; Martin, D.; Petroff, M.G.; Veiga-Lopez, A. Bisphenol S and Epidermal Growth Factor Receptor Signaling in Human Placental Cytotrophoblasts. Environ. Health Perspect. 2021, 129, 27005. [Google Scholar] [CrossRef]
- Fowden, A.L.; Moore, T. Maternal-fetal resource allocation: Co-operation and conflict. Placenta 2012, 33 (Suppl. S2), e11–e15. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Cox, B.J. Cellular analysis of trophoblast and placenta. Placenta 2017, 59 (Suppl. S1), S2–S7. [Google Scholar] [CrossRef]
- Loke, Y.W.; King, A. Immunology of human implantation: An evolutionary perspective. Hum. Reprod. 1996, 11, 283–286. [Google Scholar] [CrossRef] [Green Version]
- James, J.L.; Stone, P.R.; Chamley, L.W. The isolation and characterization of a population of extravillous trophoblast progenitors from first trimester human placenta. Hum. Reprod. 2007, 22, 2111–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Liu, D.; Diao, Z.; Wang, Z.; Ding, H.; Cai, B.; Hu, Y.; Zhao, G.; Zheng, M. Down-regulation of B2R contributes to preeclampsia by inhibiting human trophoblast cell invasion and angiogenesis. Pregnancy Hypertens. 2020, 21, 14–22. [Google Scholar] [CrossRef]
- Zhong, T.; Chen, J.; Ling, Y.; Yang, B.; Xie, X.; Yu, D.; Zhang, D.; Ouyang, J.; Kuang, H. Down-Regulation of Neuropathy Target Esterase in Preeclampsia Placenta Inhibits Human Trophoblast Cell Invasion via Modulating MMP-9 Levels. Cell Physiol. Biochem. 2018, 45, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Dackor, J.; Strunk, K.E.; Wehmeyer, M.M.; Threadgill, D.W. Altered trophoblast proliferation is insufficient to account for placental dysfunction in Egfr null embryos. Placenta 2007, 28, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Filla, M.S.; Kaul, K.L. Relative expression of epidermal growth factor receptor in placental cytotrophoblasts and choriocarcinoma cell lines. Placenta 1997, 18, 17–27. [Google Scholar] [CrossRef]
- Ko, B.K.; Lee, S.Y.; Lee, Y.H.; Hwang, I.S.; Persson, H.; Rockberg, J.; Borrebaeck, C.; Park, D.; Kim, K.T.; Uhlen, M.; et al. Combination of novel HER2-targeting antibody 1E11 with trastuzumab shows synergistic antitumor activity in HER2-positive gastric cancer. Mol. Oncol. 2015, 9, 398–408. [Google Scholar] [CrossRef]
- Faxen, M.; Nasiell, J.; Blanck, A.; Nisell, H.; Lunell, N.O. Altered mRNA expression pattern of placental epidermal growth factor receptor (EGFR) in pregnancies complicated by preeclampsia and/or intrauterine growth retardation. Am. J. Perinatol. 1998, 15, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.J.; Hsu, S.L.; Wen, M.C.; Ho, E.S.; Chou, M.M. Expression of epidermal growth factor receptor and c-erbB-2 oncoprotein in trophoblast populations of placenta accreta. Am. J. Obs. Gynecol. 2004, 191, 2106–2113. [Google Scholar] [CrossRef]
- Gupta, S.K.; Malhotra, S.S.; Malik, A.; Verma, S.; Chaudhary, P. Cell Signaling Pathways Involved During Invasion and Syncytialization of Trophoblast Cells. Am. J. Reprod. Immunol. 2016, 75, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Dilly, M.; Hambruch, N.; Haeger, J.D.; Pfarrer, C. Epidermal growth factor (EGF) induces motility and upregulates MMP-9 and TIMP-1 in bovine trophoblast cells. Mol. Reprod. Dev. 2010, 77, 622–629. [Google Scholar] [CrossRef]
- Sauer, S.J.; Tarpley, M.; Shah, I.; Save, A.V.; Lyerly, H.K.; Patierno, S.R.; Williams, K.P.; Devi, G.R. Bisphenol A activates EGFR and ERK promoting proliferation, tumor spheroid formation and resistance to EGFR pathway inhibition in estrogen receptor-negative inflammatory breast cancer cells. Carcinogenesis 2017, 38, 252–260. [Google Scholar] [CrossRef]
- Hardesty, J.E.; Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Shi, H.; Jin, J.; Vivace, B.J.; Ceresa, B.P.; Prough, R.A.; Cave, M.C. Epidermal Growth Factor Receptor Signaling Disruption by Endocrine and Metabolic Disrupting Chemicals. Toxicol. Sci. 2018, 162, 622–634. [Google Scholar] [CrossRef]
- Bass, K.E.; Morrish, D.; Roth, I.; Bhardwaj, D.; Taylor, R.; Zhou, Y.; Fisher, S.J. Human cytotrophoblast invasion is up-regulated by epidermal growth factor: Evidence that paracrine factors modify this process. Dev. Biol. 1994, 164, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Shamshirsaz, A.A.; Fox, K.A.; Erfani, H.; Clark, S.L.; Hui, S.K.; Shamshirsaz, A.A.; Rezaei, A.; Nassr, A.A.; Lake, Y.N.; Teruya, J.; et al. Coagulopathy in surgical management of placenta accreta spectrum. Eur. J. Obs. Gynecol. Reprod. Biol. 2019, 237, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sako, Y.; Minoghchi, S.; Yanagida, T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000, 2, 168–172. [Google Scholar] [CrossRef]
- Lyall, R.M.; Zilberstein, A.; Gazit, A.; Gilon, C.; Levitzki, A.; Schlessinger, J. Tyrphostins inhibit epidermal growth factor (EGF)-receptor tyrosine kinase activity in living cells and EGF-stimulated cell proliferation. J. Biol. Chem. 1989, 264, 14503–14509. [Google Scholar] [CrossRef]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey—from activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Basak, S.; Srinivas, V.; Duttaroy, A.K. Bisphenol-A impairs cellular function and alters DNA methylation of stress pathway genes in first trimester trophoblast cells. Reprod. Toxicol. 2018, 82, 72–79. [Google Scholar] [CrossRef]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef]
- Denys, H.; Derycke, L.; Hendrix, A.; Westbroek, W.; Gheldof, A.; Narine, K.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential impact of TGF-beta and EGF on fibroblast differentiation and invasion reciprocally promotes colon cancer cell invasion. Cancer Lett. 2008, 266, 263–274. [Google Scholar] [CrossRef]
- Pu, Y.; Gingrich, J.; Veiga-Lopez, A. A 3-dimensional microfluidic platform for modeling human extravillous trophoblast invasion and toxicological screening. Lab Chip 2021, 21, 546–557. [Google Scholar] [CrossRef]
- Lim, K.H.; Zhou, Y.; Janatpour, M.; McMaster, M.; Bass, K.; Chun, S.H.; Fisher, S.J. Human cytotrophoblast differentiation/invasion is abnormal in pre-eclampsia. Am. J. Pathol. 1997, 151, 1809–1818. [Google Scholar]
- Rantakokko, P.; Main, K.M.; Wohlfart-Veje, C.; Kiviranta, H.; Airaksinen, R.; Vartiainen, T.; Skakkebaek, N.E.; Toppari, J.; Virtanen, H.E. Association of placenta organotin concentrations with growth and ponderal index in 110 newborn boys from Finland during the first 18 months of life: A cohort study. Environ. Health Glob. Access Sci. Source 2014, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, G.; Pussetto, M.; Rose, M.; Staff, A.C.; Blois, S.M.; Toblli, J.E. Defective trophoblast invasion underlies fetal growth restriction and preeclampsia-like symptoms in the stroke-prone spontaneously hypertensive rat. Mol. Hum. Reprod. 2017, 23, 509–519. [Google Scholar] [CrossRef]
- Sebire, N.J.; Fox, H.; Backos, M.; Rai, R.; Paterson, C.; Regan, L. Defective endovascular trophoblast invasion in primary antiphospholipid antibody syndrome-associated early pregnancy failure. Hum. Reprod. 2002, 17, 1067–1071. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kannan, K.; Tan, H.; Zheng, Z.; Feng, Y.L.; Wu, Y.; Widelka, M. Bisphenol Analogues Other Than BPA: Environmental Occurrence, Human Exposure, and Toxicity-A Review. Environ. Sci. Technol. 2016, 50, 5438–5453. [Google Scholar] [CrossRef] [PubMed]
- Sol, C.M.; van Zwol-Janssens, C.; Philips, E.M.; Asimakopoulos, A.G.; Martinez-Moral, M.P.; Kannan, K.; Jaddoe, V.W.V.; Trasande, L.; Santos, S. Maternal bisphenol urine concentrations, fetal growth and adverse birth outcomes: A population-based prospective cohort. Environ. Health Glob. Access Sci. Source 2021, 20, 60. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, H.; Braun, J.M.; Zheng, T.; Zhang, B.; Xia, W.; Zhang, W.; Li, J.; Zhou, Y.; Li, H.; et al. Associations of Trimester-Specific Exposure to Bisphenols with Size at Birth: A Chinese Prenatal Cohort Study. Environ. Health Perspect. 2019, 127, 107001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, S.; Duttaroy, A.K. Leptin induces tube formation in first-trimester extravillous trophoblast cells. Eur. J. Obs. Gynecol. Reprod. Biol. 2012, 164, 24–29. [Google Scholar] [CrossRef]
- Gingrich, J.; Pu, Y.; Upham, B.L.; Hulse, M.; Pearl, S.; Martin, D.; Avery, A.; Veiga-Lopez, A. Bisphenol S enhances gap junction intercellular communication in ovarian theca cells. Chemosphere 2021, 263, 128304. [Google Scholar] [CrossRef]
- Poteser, M.; Hutter, H.P.; Moshammer, H.; Weitensfelder, L. Perfluoroctanoic acid (PFOA) enhances NOTCH-signaling in an angiogenesis model of placental trophoblast cells. Int. J. Hyg. Environ. Health 2020, 229, 113566. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Iwano, T.; Takeda, S. Estrogen and EGFR Pathways Regulate Notch Signaling in Opposing Directions for Multi-Ciliogenesis in the Fallopian Tube. Cells 2019, 8, 933. [Google Scholar] [CrossRef] [Green Version]
- Marroqui, L.; Martinez-Pinna, J.; Castellano-Munoz, M.; Dos Santos, R.S.; Medina-Gali, R.M.; Soriano, S.; Quesada, I.; Gustafsson, J.A.; Encinar, J.A.; Nadal, A. Bisphenol-S and Bisphenol-F alter mouse pancreatic beta-cell ion channel expression and activity and insulin release through an estrogen receptor ERbeta mediated pathway. Chemosphere 2021, 265, 129051. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, J.; Albitar, L.; Laidler, L.L.; Abdullah, R.; Leslie, K.K. Blocking Epidermal Growth Factor Receptor Signaling in HTR-8/SVneo First Trimester Trophoblast Cells Results in Dephosphorylation of PKBalpha/AKT and Induces Apoptosis. Obs. Gynecol. Int. 2011, 2011, 896896. [Google Scholar]
- Ahmann, F.R.; Garewal, H.S.; Schifman, R.; Celniker, A.; Rodney, S. Intracellular adenosine triphosphate as a measure of human tumor cell viability and drug modulated growth. In Vitro Cell. Dev. Biol. 1987, 23, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Sevin, B.U.; Peng, Z.L.; Perras, J.P.; Ganjei, P.; Penalver, M.; Averette, H.E. Application of an ATP-bioluminescence assay in human tumor chemosensitivity testing. Gynecol. Oncol. 1988, 31, 191–204. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ticiani, E.; Pu, Y.; Gingrich, J.; Veiga-Lopez, A. Bisphenol S Impairs Invasion and Proliferation of Extravillous Trophoblasts Cells by Interfering with Epidermal Growth Factor Receptor Signaling. Int. J. Mol. Sci. 2022, 23, 671. https://doi.org/10.3390/ijms23020671
Ticiani E, Pu Y, Gingrich J, Veiga-Lopez A. Bisphenol S Impairs Invasion and Proliferation of Extravillous Trophoblasts Cells by Interfering with Epidermal Growth Factor Receptor Signaling. International Journal of Molecular Sciences. 2022; 23(2):671. https://doi.org/10.3390/ijms23020671
Chicago/Turabian StyleTiciani, Elvis, Yong Pu, Jeremy Gingrich, and Almudena Veiga-Lopez. 2022. "Bisphenol S Impairs Invasion and Proliferation of Extravillous Trophoblasts Cells by Interfering with Epidermal Growth Factor Receptor Signaling" International Journal of Molecular Sciences 23, no. 2: 671. https://doi.org/10.3390/ijms23020671
APA StyleTiciani, E., Pu, Y., Gingrich, J., & Veiga-Lopez, A. (2022). Bisphenol S Impairs Invasion and Proliferation of Extravillous Trophoblasts Cells by Interfering with Epidermal Growth Factor Receptor Signaling. International Journal of Molecular Sciences, 23(2), 671. https://doi.org/10.3390/ijms23020671