
Structural and Functional Characterization of Four Novel Fibrinogen Mutations in FGB Causing Congenital Fibrinogen Disorder

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Coagulation Screening Tests of Patients with CFD Suspicion and Genetic Analysis
2.2. Characterization of Each Patient’s Fibrinogen Behavior
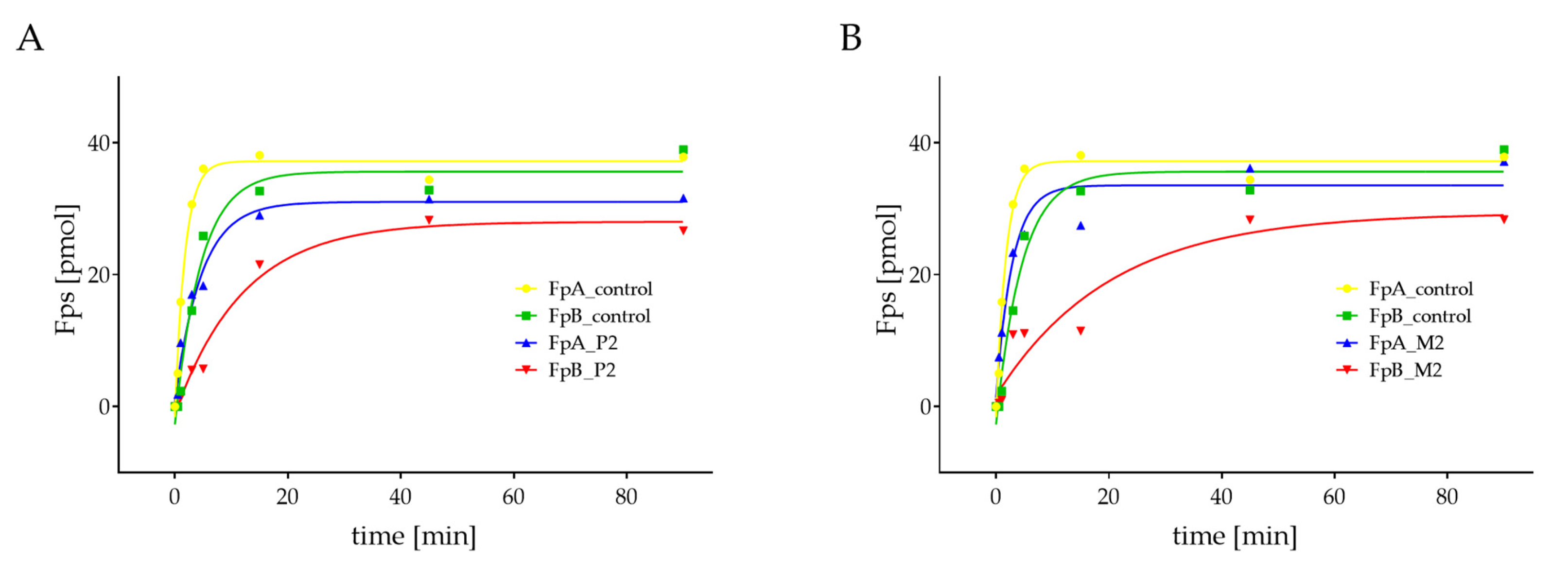
2.3. Quantification of Thrombin-Catalyzed Cleavage of Fps
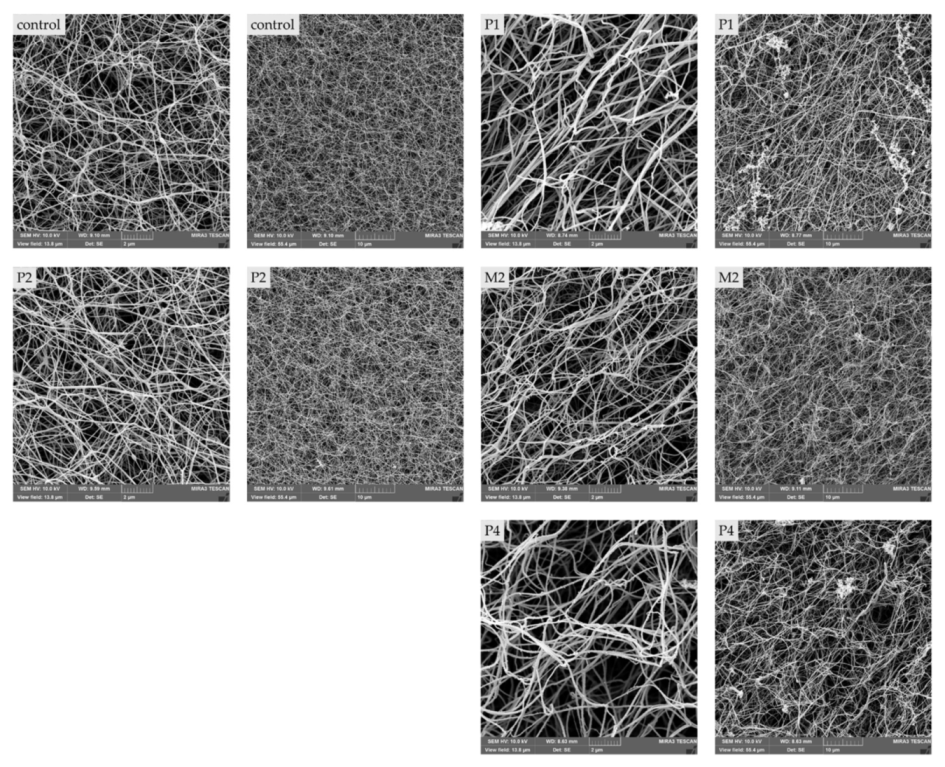
2.4. Morphologic Differences of Patient’S Fibrin Clots Compared to Controls
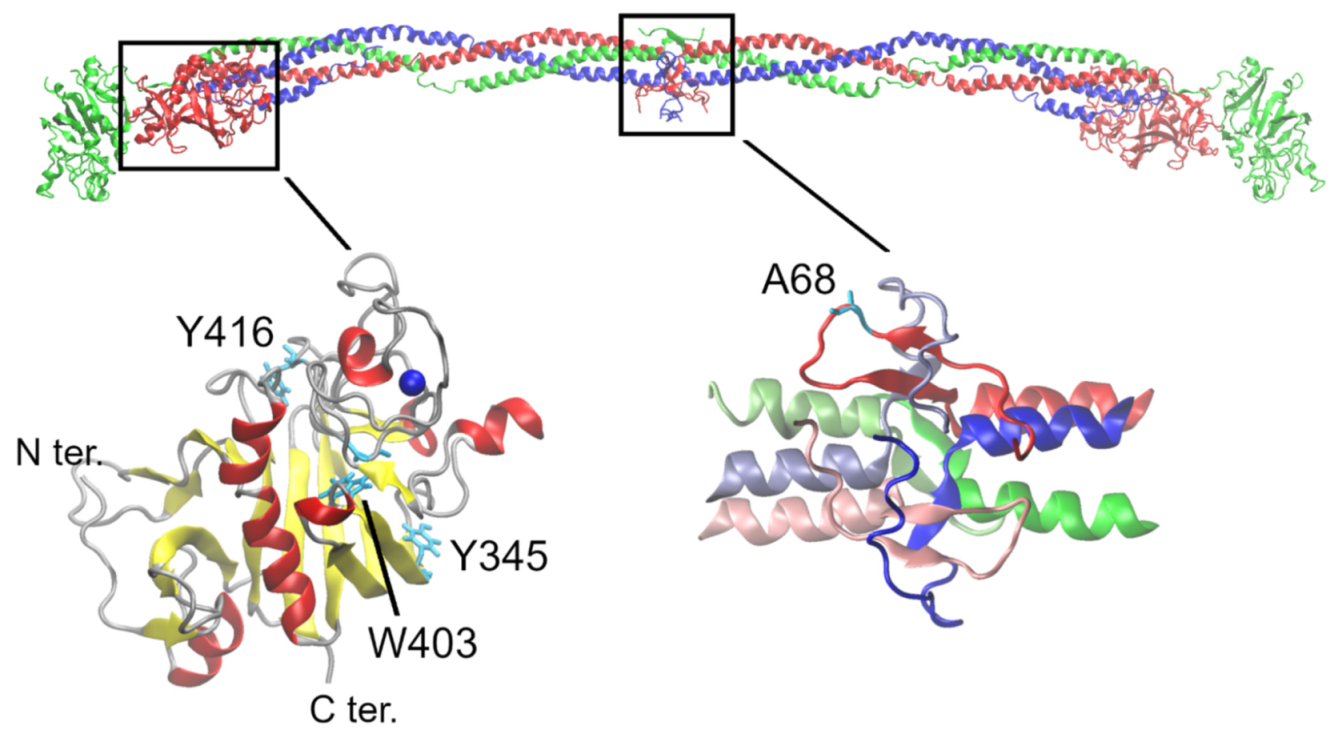
2.5. Protein Modeling of BβA68S
3. Discussion
3.1. Molecular Characteristic of 4 Novel Fibrinogen Variants
3.2. Correlation between Mutations, Fibrinogen Function, Clot Structures, and Clinical Phenotype
3.2.1. Fibrinogen Karlovy Vary and Fibrinogen Most
3.2.2. Fibrinogen Svetec
3.3. Homology Model of Fibrinogen Svetec
4. Materials and Methods
4.1. Blood Collection and Coagulation Screening Tests
4.2. Fibrinogen DNA Sequencing
4.3. Fibrinogen Purification
4.4. Fibrinogen Polymerization and Fibrinolysis
4.5. SEM
4.6. Quantification of Fps Cleavage by RP-HPLC
4.7. Statistical Analysis
4.8. Homology Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henschen, A.; Mcdonagh, J. Chapter 7 Fibrinogen, fibrin and factor XIII. New Compr. Biochem. 1986, 13, 171–241. [Google Scholar] [CrossRef]
- Fowler, W.E.; Erickson, H.P. Trinodular structure of fibrinogen: Confirmation by both shadowing and negative stain electron microscopy. J. Mol. Biol. 1979, 134, 241–249. [Google Scholar] [CrossRef]
- Medved, L.; Weisel, J.W. Fibrinogen; of Scientific Standardization Committee of International Society on Thrombosis; Haemostasis Recommendations for nomenclature on fibrinogen and fibrin. J. Thromb. Haemost. 2009, 7, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Undas, A.; Palla, R.; Thachil, J.; de Moerloose, P. Diagnosis and classification of congenital fibrinogen disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1887–1890. [Google Scholar] [CrossRef]
- Casini, A.; Neerman-Arbez, M.; Ariëns, R.A.; de Moerloose, P. Dysfibrinogenemia: From molecular anomalies to clinical manifestations and management. J. Thromb. Haemost. 2015, 13, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanss, M.; Biot, F. A database for human fibrinogen variants. Ann. N. Y. Acad. Sci. 2001, 936, 89–90. [Google Scholar] [CrossRef]
- Casini, A.; Lukowski, S.; Quintard, V.L.; Crutu, A.; Zak, M.; Regazzoni, S.; De Moerloose, P.; Neerman-Arbez, M. FGB mutations leading to congenital quantitative fibrinogen deficiencies: An update and report of four novel mutations. Thromb. Res. 2014, 133, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Di Sanza, C.; Caille, D.; de Moerloose, P.; Scheib, H.; Meda, P.; Neerman-Arbez, M. Quality control of fibrinogen secretion in the molecular pathogenesis of congenital afibrinogenemia. Hum. Mol. Genet. 2005, 14, 3271–3280. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; De Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [CrossRef]
- Sumitha, E.; Jayandharan, G.R.; Arora, N.; Abraham, A.; David, S.; Devi, G.S.; Shenbagapriya, P.; Nair, S.C.; George, B.; Mathews, V.; et al. Molecular basis of quantitative fibrinogen disorders in 27 patients from India. Haemophilia 2013, 19, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Vilar, R.; Beauverd, Y.; Aslan, D.; Devreese, K.; Mondelaers, V.; Alberio, L.; Gubert, C.; de Moerloose, P.; Neerman-Arbez, M. Protein modelling to understand FGB mutations leading to congenital hypofibrinogenaemia. Haemophilia 2017, 23, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Koopman, J.; Haverkate, F.; Lord, S.; Grimbergen, J.; Mannucci, P. Molecular basis of fibrinogen Naples associated with defective thrombin binding and thrombophilia. Homozygous substitution of B beta 68 Ala—Thr. J. Clin. Investig. 1992, 90, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ding, Q.; Chen, Y.; Ouyang, Q.; Jiang, L.; Dai, J.; Lu, Y.; Wu, X.; Liang, Q.; Wang, H.; et al. Clinical features and molecular basis of 102 Chinese patients with congenital dysfibrinogenemia. Blood Cells. Mol. Dis. 2015, 55, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kibe, T.; Matsubara, R.; Koizumi, S.; Nara, K.; Amano, K.; Okumura, N. Congenital dysfibrinogenemia in a Japanese family with fibrinogen Naples (BβAla68Thr) manifesting as superior sagittal sinus thrombosis. Blood Coagul. Fibrinolysis 2017, 28, 580–584. [Google Scholar] [CrossRef]
- Meh, D.A.; Mosesson, M.W.; Siebenlist, K.R.; Simpson-Haidaris, P.J.; Brennan, S.O.; DiOrio, J.P.; Thompson, K.; Di Minno, G. Fibrinogen naples I (B beta A68T) nonsubstrate thrombin-binding capacities. Thromb. Res. 2001, 103, 63–73. [Google Scholar] [CrossRef]
- Di Minno, G.; Martinez, J.; Cirillo, F.; Cerbone, A.M.; Silver, M.J.; Colucci, M.; Margaglione, M.; Tauro, R.; Semeraro, N.; Quattrone, A.; et al. A role for platelets and thrombin in the juvenile stroke of two siblings with defective thrombin-adsorbing capacity of fibrin(ogen). Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlín, R.; Reicheltová, Z.; Sobotková, A.; Suttnar, J.; Salaj, P.; Pospíšilová, D.; Smejkal, P.; Chrastinová, L.; Dyr, J.E. Three cases of abnormal fibrinogens: Sumperk (Bbeta His67Leu), Unicov (Bbeta Gly414Ser), and Brno (gammaArg275His). Thromb. Haemost. 2008, 100, 1199–1200. [Google Scholar]
- Kotlin, R.; Reicheltova, Z.; Suttnar, J.; Salaj, P.; Hrachovinova, I.; Riedel, T.; Maly, M.; Oravec, M.; Kvasnicka, J.; Dyr, J.E. Two novel fibrinogen variants in the C-terminus of the Bbeta-chain: Fibrinogen Rokycany and fibrinogen Znojmo. J. Thromb. Thrombolysis 2010, 30, 311–318. [Google Scholar] [CrossRef]
- Duga, S.; Asselta, R.; Santagostino, E.; Zeinali, S.; Simonic, T.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. Missense mutations in the human β fibrinogen gene cause congenital afibrinogenemia by impairing fibrinogen secretion. Blood 2000, 95, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Hanss, M.; Ffrench, P.; Vinciguerra, C.; Bertrands, M.A.; De Mazancourt, P. Four cases of hypofibrinogenemia associated with four novel mutations. J. Thromb. Haemost. 2005, 3, 2347–2349. [Google Scholar] [CrossRef]
- Aung, N.N.; Kennedy, H.; Faed, J.M.; Brennan, S.O. Novel heterozygous Bbeta (c.1311T>A) mutation (Fibrinogen St Kilda) associated with recurrent pregnancy loss. Pathology 2015, 47, 583–585. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Redman, C.M. Identification of B beta chain domains involved in human fibrinogen assembly. J. Biol. Chem. 1992, 267, 21727–21732. [Google Scholar] [CrossRef]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A novel nonsense mutation in fgb (C.1421g>a; p.trp474ter) in the beta chain of fibrinogen causing hypofibrinogenemia with bleeding phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef]
- Carr, M.E.; Gabriel, D.A.; McDonagh, J. Influence of Ca2+ on the structure of reptilase-derived and thrombin-derived fibrin gels. Biochem. J. 1986, 239, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Nair, C.H.; Shah, G.A.; Dhall, D.P. Effect of temperature, pH and ionic strength and composition on fibrin network structure and its development. Thromb. Res. 1986, 42, 809–816. [Google Scholar] [CrossRef]
- Weisel, J.W.; Nagaswami, C. Computer modeling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: Clot structure and assembly are kinetically controlled. Biophys. J. 1992, 63, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.A.; Muga, K.; Boothroyd, E.M. The effect of fibrin structure on fibrinolysis. J. Biol. Chem. 1992, 267, 24259–24263. [Google Scholar] [CrossRef]
- Torbet, J. Fibrin assembly in human plasma and fibrinogen/albumin mixtures. Biochemistry 1986, 25, 5309–5314. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liang, M.; Yang, J.; Zhang, X. Congenital hypofibrinogenemia in pregnancy: A report of 11 cases. Blood Coagul. Fibrinolysis 2018, 29, 155–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammaritano, L.R. Antiphospholipid syndrome. Best Pract. Res. Clin. Rheumatol. 2020, 34. [Google Scholar] [CrossRef]
- Mullin, J.L.; Gorkun, O.V.; Lord, S.T. Decreased lateral aggregation of a variant recombinant fibrinogen provides insight into the polymerization mechanism. Biochemistry 2000, 39, 9843–9849. [Google Scholar] [CrossRef]
- Lord, S.T.; Strickland, E.; Jayjock, E. Strategy for recombinant multichain protein synthesis: Fibrinogen B beta-chain variants as thrombin substrates. Biochemistry 1996, 35, 2342–2348. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Nagata, K.; Taira, C.; Higuchi, Y.; Arai, S.; Okumura, N. Fibrin monomers derived from thrombogenic dysfibrinogenemia, Naples-type variant (BβAla68Thr), showed almost entirely normal polymerization. Thromb. Res. 2018, 172, 1–3. [Google Scholar] [CrossRef]
- Brennan, S.O.; Roncolato, F. Novel fibrinogen (B β401Gly→Val) presents as dys- or hypodysfibrinogenaemia due to alterations in sialic acid content. Thromb. Haemost. 2011, 106, 551–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, J.J.; Snoek, C.J.M.; Rijken, D.C.; De Maat, M.P.M. Effects of post-translational modifications of fibrinogen on clot formation, clot structure, and fibrinolysis: A systematic review. Arterioscler. Thromb. Vasc. Biol. 2019, 40, 554–569. [Google Scholar] [CrossRef]
- Clauss, A. Rapid physiological coagulation method in determination of fibrinogen. Acta Haematol. 1957, 17, 237–246. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, S.O.; Hammonds, B.; George, P.M. Aberrant hepatic processing causes removal of activation peptide and primary polymerisation site from fibrinogen Canterbury (A alpha 20 Val –> Asp). J. Clin. Investig. 1995, 96, 2854–2858. [Google Scholar] [CrossRef] [PubMed]
- Suttnar, J.; Dyr, J.E.; Fořtová, H.; Pristach, J. Determination of fibrinonopeptides by high resolution liquid chromotagraphy. Biochem. Clin. Bohemoslov. 1989, 18, 17–25. [Google Scholar]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal structure of human fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Fbg Clauss [g/L] (1.9–4.2) | Fbg Total [g/L] (1.8–4.2) | aPTT-INR (0.8–1.2) | PT-INR (0.8–1.2) | TT-INR (0.85–1.15) | RT [s] | Clinical Phenotype | Mutation |
|---|---|---|---|---|---|---|---|---|
| P1 | 1.3 | 1.7 | 1.0 | 1.1 | 1.2 | 22.4 (18.1–22.1) | A | BβY416C |
| P2 | 1.6 | 2.9 | 1.1 | 1.0 | 1.2 | 21.0 (18.9–22.9) | epistaxis | BβA68S |
| M2 | 1.8 | 2.9 | 1.0 | 1.0 | 1.2 | 20.9 (18.9–22.9) | A | BβA68S |
| B2 | 2.1 | 2.1 | 1.1 | 1.0 | 0.9 | 19.8 (18.9–22.9) | epistaxis | none |
| P3 | <0.2 | 0.0 | >6 | NA | >12 | >300 (17.2–21.2) | A | BβY345* |
| M3 | 2.0 | 2.0 | 1.5 | 1.1 | 1.0 | 17.4 (17.2–21.2) | A | NA |
| F3 | 1.8 | 2.0 | 1.0 | 1.1 | 0.9 | 16.7 (17.2–21.2) | A | NA |
| P4 | 1.6 | 1.7 | NA | NA | 1.0 | 18.6 (18.8–22.8) | pulmonary embolism | BβW403* |
| Control | P1 | P2 | M2 | P4 | |
|---|---|---|---|---|---|
| Plasma (1 g/L) | |||||
| lag time [s] | 140 | 340 | 1940 | 300 | NA |
| maxOD | 10.6 | 6.4 | 1.3 | 10.5 | NA |
| slope (maxOD/s × 103) | 12.0 | 2.8 | 0.5 | 12.8 | NA |
| T90% [s] | 420 | 1640 | 2360 | 540 | NA |
| Fibrinolysis | |||||
| T50% [s] | 240 | 300 | 360 | 340 | NA |
| T100% [s] | 560 | 1700 | UD | 1880 | NA |
| Purified Fbg (1 g/L) | |||||
| lag time [s] | 270 | 460 | 960 | 1360 | 240 |
| maxOD | 10.1 | 2.8 | 3.1 | 2.2 | 3.0 |
| slope (maxOD/s × 103) | 6.2 | 1.5 | 1.3 | 0.9 | 1.8 |
| T90% [s] | 1000 | 1240 | 2280 | 2300 | 880 |
| Fibrinolysis | |||||
| T50% [s] | 160 | 280 | 180 | 180 | 300 |
| T100% [s] | 1380 | 1480 | 1420 | 1160 | 1780 |
| Patient | Fiber Thickness [nm] | Average No. of Fibers per 1 µm2 |
|---|---|---|
| control | 78.0 ± 22.6 | 15.0 ± 2.6 |
| P1 | 109.4 ± 33.8 | 9.5 ± 2.6 |
| P2 | 79.6 ± 23.5 | 16.7 ± 2.6 |
| M2 | 88.0 ± 27.2 | 12.1 ± 2.9 |
| P4 | 118.8 ± 34.5 | 6.7 ± 2.0 |
| p-Value | p < 0.0001 | p < 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceznerová, E.; Kaufmanová, J.; Sovová, Ž.; Štikarová, J.; Loužil, J.; Kotlín, R.; Suttnar, J. Structural and Functional Characterization of Four Novel Fibrinogen Mutations in FGB Causing Congenital Fibrinogen Disorder. Int. J. Mol. Sci. 2022, 23, 721. https://doi.org/10.3390/ijms23020721
Ceznerová E, Kaufmanová J, Sovová Ž, Štikarová J, Loužil J, Kotlín R, Suttnar J. Structural and Functional Characterization of Four Novel Fibrinogen Mutations in FGB Causing Congenital Fibrinogen Disorder. International Journal of Molecular Sciences. 2022; 23(2):721. https://doi.org/10.3390/ijms23020721
Chicago/Turabian StyleCeznerová, Eliška, Jiřina Kaufmanová, Žofie Sovová, Jana Štikarová, Jan Loužil, Roman Kotlín, and Jiří Suttnar. 2022. "Structural and Functional Characterization of Four Novel Fibrinogen Mutations in FGB Causing Congenital Fibrinogen Disorder" International Journal of Molecular Sciences 23, no. 2: 721. https://doi.org/10.3390/ijms23020721
APA StyleCeznerová, E., Kaufmanová, J., Sovová, Ž., Štikarová, J., Loužil, J., Kotlín, R., & Suttnar, J. (2022). Structural and Functional Characterization of Four Novel Fibrinogen Mutations in FGB Causing Congenital Fibrinogen Disorder. International Journal of Molecular Sciences, 23(2), 721. https://doi.org/10.3390/ijms23020721