
Tilting the Balance: Therapeutic Prospects of CD83 as a Checkpoint Molecule Controlling Resolution of Inflammation

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. CD83: From Maturation Marker to Pro-Resolving Checkpoint Molecule
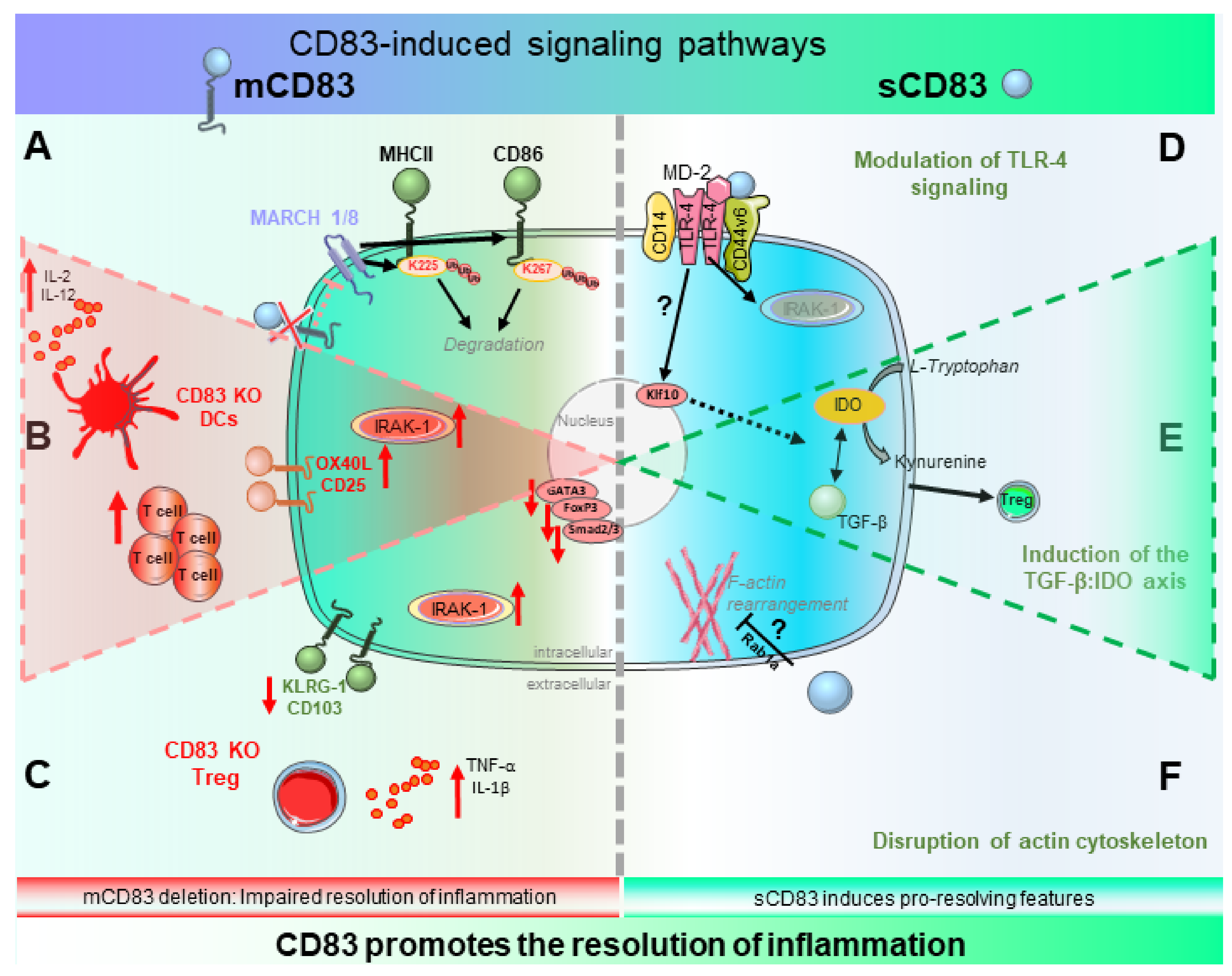
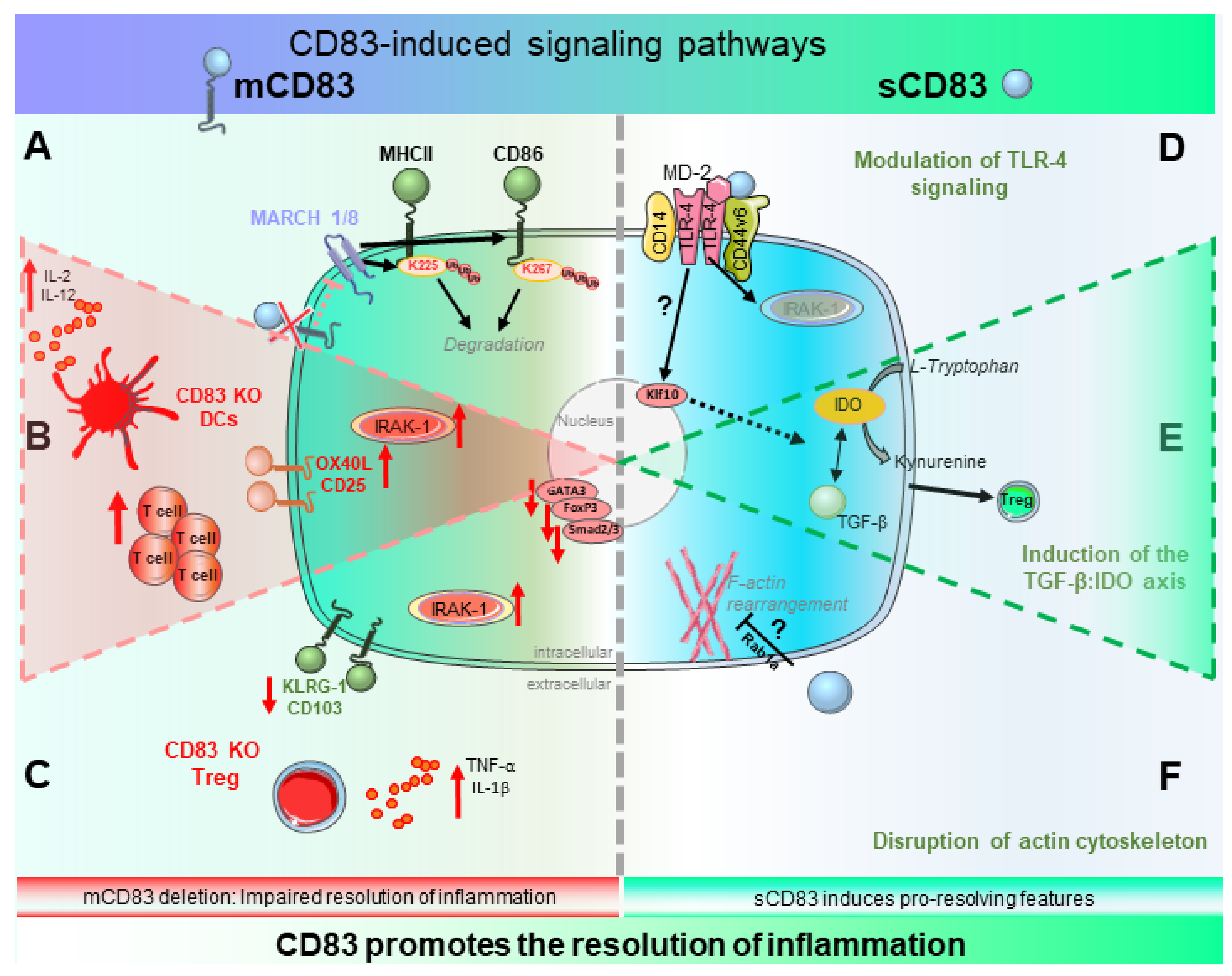
2.1. Biological Function of CD83 and Its Induced Signaling Events
2.1.1. Biological Relevance of mCD83—Beware the Ides of MARCH
2.1.2. Signaling of sCD83—A Tale of Two Pathways
2.2. Role of mCD83 in the Resolution of Inflammation
2.2.1. CD83 Expression by DCs—Fine-Tuning of Inflammation and Its Resolution
2.2.2. CD83 Expression by Tregs—Gate-Keeper of Tolerance
3. Clinical Relevance of sCD83 for Therapeutic Purposes
3.1. sCD83 Promotes the Resolution of Chronic Inflammation
3.1.1. Autoimmune Disorders
3.1.2. Inflammatory Bowel Disease
3.1.3. Rheumatoid Arthritis
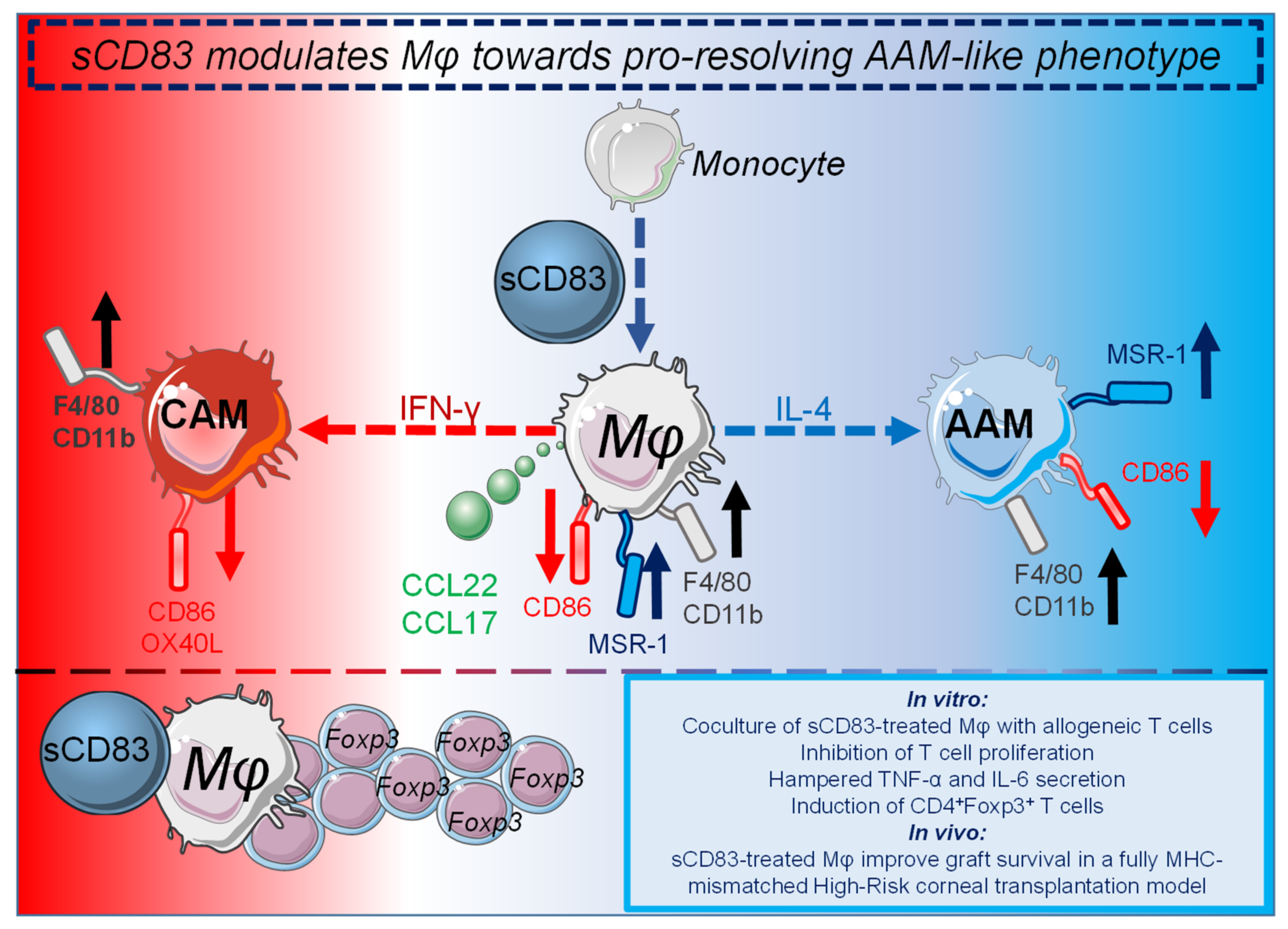
3.2. sCD83 Prevents Graft Rejection by Induction of Tolerogenic Mechanisms
4. sCD83: Conclusions, New Insights and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lammermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rius, A.; Desai, A.; Yuen, D.; Johnston, A.P.R.; Voelcker, N.H. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat. Nanotechnol. 2021, 16, 37–46. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Filep, J.G.; Ariel, A. Neutrophil heterogeneity and fate in inflaMed. tissues: Implications for the resolution of inflammation. Am. J. Physiol Cell Physiol. 2020, 319, C510–C532. [Google Scholar] [CrossRef]
- Marwick, J.A.; Mills, R.; Kay, O.; Michail, K.; Stephen, J.; Rossi, A.G.; Dransfield, I.; Hirani, N. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-kappaB activation. Cell Death Dis. 2018, 9, 665. [Google Scholar] [CrossRef]
- Munoz-Rojas, A.R.; Kelsey, I.; Pappalardo, J.L.; Chen, M.; Miller-Jensen, K. Co-stimulation with opposing macrophage polarization cues leads to orthogonal secretion programs in individual cells. Nat. Commun. 2021, 12, 301. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, F.; Vigne, S.; Pot, C. Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 2019, 41, 711–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schett, G. Resolution of inflammation in arthritis. Semin. Immunopathol. 2019, 41, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Mori, D.N.; Kreisel, D.; Fullerton, J.N.; Gilroy, D.W.; Goldstein, D.R. Inflammatory triggers of acute rejection of organ allografts. Immunol. Rev. 2014, 258, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Schwarting, R.; Smith, H.M.; Tedder, T.F. A Novel Cell-Surface Molecule Expressed by Human Interdigitating Reticulum Cells, Langerhans Cells, and Activated Lymphocytes Is a New Member of the Ig Superfamily. J. Immunol. 1992, 149, 735–742. [Google Scholar]
- Grosche, L.; Knippertz, I.; Konig, C.; Royzman, D.; Wild, A.B.; Zinser, E.; Sticht, H.; Muller, Y.A.; Steinkasserer, A.; Lechmann, M. The CD83 Molecule—An Important Immune Checkpoint. Front. Immunol. 2020, 11, 721. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ju, X.; Silveira, P.A.; Abadir, E.; Hsu, W.H.; Hart, D.N.J.; Clark, G.J. CD83: Activation Marker for Antigen Presenting Cells and Its Therapeutic Potential. Front. Immunol. 2019, 10, 1312. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Berchtold, S.; Muhl-Zurbes, P.; Heufler, C.; Winklehner, P.; Schuler, G.; Steinkasserer, A. Cloning, recombinant expression and biochemical characterization of the murine CD83 molecule which is specifically upregulated during dendritic cell maturation. FEBS Lett. 1999, 461, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Hock, B.D.; Kato, M.; McKenzie, J.L.; Hart, D.N. A soluble form of CD83 is released from activated dendritic cells and B lymphocytes, and is detectable in normal human sera. Int. Immunol. 2001, 13, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudziak, D.; Nimmerjahn, F.; Bornkamm, G.W.; Laux, G. Alternative splicing generates putative soluble CD83 proteins that inhibit T cell proliferation. J. Immunol. 2005, 174, 6672–6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hock, B.D.; Haring, L.F.; Steinkasserer, A.; Taylor, K.G.; Patton, W.N.; McKenzie, J.L. The soluble form of CD83 is present at elevated levels in a number of hematological malignancies. Leukemia Res. 2004, 28, 237–241. [Google Scholar] [CrossRef]
- Karampoor, S.; Zahednasab, H.; Etemadifar, M.; Keyvani, H. The levels of soluble forms of CD21 and CD83 in multiple sclerosis. J. NeuroImmunol. 2018, 320, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Hock, B.D.; O’Donnell, J.L.; Taylor, K.; Steinkasserer, A.; McKenzie, J.L.; Rothwell, A.G.; Summers, K.L. Levels of the soluble forms of CD80, CD86, and CD83 are elevated in the synovial fluid of rheumatoid arthritis patients. Tissue Antigens 2006, 67, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Lechmann, M.; Berchtold, S.; Hauber, J.; Steinkasserer, A. CD83 on dendritic cells: More than just a marker for maturation. Trends Immunol. 2002, 23, 273–275. [Google Scholar] [CrossRef]
- Bates, J.M.; Flanagan, K.; Mo, L.; Ota, N.; Ding, J.; Ho, S.; Liu, S.; Roose-Girma, M.; Warming, S.; Diehl, L. Dendritic cell CD83 homotypic interactions regulate inflammation and promote mucosal homeostasis. Mucosal. Immunol. 2015, 8, 414–428. [Google Scholar] [CrossRef]
- Wild, A.B.; Krzyzak, L.; Peckert, K.; Stich, L.; Kuhnt, C.; Butterhof, A.; Seitz, C.; Mattner, J.; Gruner, N.; Gansbauer, M.; et al. CD83 orchestrates immunity toward self and non-self in dendritic cells. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Tze, L.E.; Horikawa, K.; Domaschenz, H.; Howard, D.R.; Roots, C.M.; Rigby, R.J.; Way, D.A.; Ohmura-Hoshino, M.; Ishido, S.; Andoniou, C.E.; et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 2011, 208, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, S.; Wang, J.M.; Yang, D.; Gong, W.H.; Kamohara, H.; Yoshimura, T. Expression of CCR6 and CD83 by cytokine-activated human neutrophils. Blood 2000, 96, 3958–3963. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Lee, S.H.; Lu, J. CD83 is preforMed. inside monocytes, macrophages and dendritic cells, but it is only stably expressed on activated dendritic cells. Biochem. J. 2005, 385, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.O.; Trumpfheller, C.; Mehlhoop, U.; More, S.; Fleischer, B.; von Bonin, A. Activation-induced expression of murine CD83 on T cells and identification of a specific CD83 ligand on murine B cells. Int. Immunol. 2000, 12, 1347–1351. [Google Scholar] [CrossRef] [Green Version]
- Mailliard, R.B.; Alber, S.M.; Shen, H.; Watkins, S.C.; Kirkwood, J.M.; Herberman, R.B.; Kalinski, P. IL-18-induced CD83+CCR7+ NK helper cells. J. Exp. Med. 2005, 202, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Doebbeler, M.; Koenig, C.; Krzyzak, L.; Seitz, C.; Wild, A.; Ulas, T.; Bassler, K.; Kopelyanskiy, D.; Butterhof, A.; Kuhnt, C.; et al. CD83 expression is essential for Treg cell differentiation and stability. JCI Insight 2018, 3, e99712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Rohrscheidt, J.; Petrozziello, E.; Nedjic, J.; Federle, C.; Krzyzak, L.; Ploegh, H.L.; Ishido, S.; Steinkasserer, A.; Klein, L. Thymic CD4 T cell selection requires attenuation of March8-mediated MHCII turnover in cortical epithelial cells through CD83. J. Exp. Med. 2016, 213, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jin, Q.R.; Mi, Y.; Liu, J.Q.; Liu, Z.Q.; Wang, S.; Liu, Z.G.; Yang, P.C.; Zheng, P.Y. Intestinal Epithelial Cell-Derived CD83 Contributes to Regulatory T-Cell Generation and Inhibition of Food Allergy. J. Innate Immun. 2021, 13, 295–305. [Google Scholar] [CrossRef]
- Mo, L.H.; Luo, X.Q.; Yang, G.; Liu, J.Q.; Yang, L.T.; Liu, Z.Q.; Wang, S.; Liu, D.B.; Liu, Z.G.; Yang, P.C. Epithelial cell-derived CD83 restores immune tolerance in the airway mucosa by inducing regulatory T-cell differentiation. Immunology 2021, 163, 310–322. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Muhl-Zurbes, P.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J. Gen. Virol. 2014, 95, 1366–1375. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Grosche, L.; Kummer, M.; Muhl-Zurbes, P.; Kamm, L.; Scherer, M.; Latzko, M.; Stamminger, T.; Steinkasserer, A. The Major Immediate-Early Protein IE2 of Human Cytomegalovirus Is Sufficient to Induce Proteasomal Degradation of CD83 on Mature Dendritic Cells. Front. Microbiol. 2017, 8, 119. [Google Scholar] [CrossRef]
- Grosche, L.; Muhl-Zurbes, P.; Ciblis, B.; Krawczyk, A.; Kuhnt, C.; Kamm, L.; Steinkasserer, A.; Heilingloh, C.S. Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells. Viruses 2020, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Ju, X.; Lee, K.; Clarke, C.; Hsu, J.L.; Abadir, E.; Bryant, C.E.; Pears, S.; Sunderland, N.; Heffernan, S.; et al. CD83 is a new potential biomarker and therapeutic target for Hodgkin lymphoma. Haematologica 2018, 103, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, Y.; Tu, L.; Miller, A.S.; Bock, C.; Fujimoto, M.; Doyle, C.; Steeber, D.A.; Tedder, T.F. CD83 expression influences CD4+ T cell development in the thymus. Cell 2002, 108, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Zinser, E.; Lechmann, M.; Golka, A.; Lutz, M.B.; Steinkasserer, A. Prevention and treatment of experimental autoimmune encephalomyelitis by soluble CD83. J. Exp. Med. 2004, 200, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Starke, C.; Steinkasserer, A.; Voll, R.E.; Zinser, E. Soluble human CD83 ameliorates lupus in NZB/W F1 mice. Immunobiology 2013, 218, 1411–1415. [Google Scholar] [CrossRef]
- Eckhardt, J.; Kreiser, S.; Dobbeler, M.; Nicolette, C.; DeBenedette, M.A.; Tcherepanova, I.Y.; Ostalecki, C.; Pommer, A.J.; Becker, C.; Gunther, C.; et al. Soluble CD83 ameliorates experimental colitis in mice. Mucosal. Immunol. 2014, 7, 1006–1018. [Google Scholar] [CrossRef]
- Lin, W.; Man, X.; Li, P.; Song, N.; Yue, Y.; Li, B.; Li, Y.; Sun, Y.; Fu, Q. NK cells are negatively regulated by sCD83 in experimental autoimmune uveitis. Sci. Rep. 2017, 7, 12895. [Google Scholar] [CrossRef]
- Lin, W.; Buscher, K.; Wang, B.; Fan, Z.; Song, N.; Li, P.; Yue, Y.; Li, B.; Li, C.; Bi, H. Soluble CD83 Alleviates Experimental Autoimmune Uveitis by Inhibiting Filamentous Actin-Dependent Calcium Release in Dendritic Cells. Front. Immunol. 2018, 9, 1567. [Google Scholar] [CrossRef]
- McHugh, J. Soluble CD83: A proresolving mediator in inflammatory arthritis? Nat. Rev. Rheumatol. 2019, 15, 319. [Google Scholar] [CrossRef]
- Royzman, D.; Andreev, D.; Stich, L.; Rauh, M.; Bauerle, T.; Ellmann, S.; Boon, L.; Kindermann, M.; Peckert, K.; Bozec, A.; et al. Soluble CD83 Triggers Resolution of Arthritis and Sustained Inflammation Control in IDO Dependent Manner. Front. Immunol. 2019, 10, 633. [Google Scholar] [CrossRef]
- Kretschmer, B.; Luthje, K.; Ehrlich, S.; Osterloh, A.; Piedavent, M.; Fleischer, B.; Breloer, M. CD83 on murine APC does not function as a costimulatory receptor for T cells. Immunol. Lett. 2008, 120, 87–95. [Google Scholar] [CrossRef]
- Kuwano, Y.; Prazma, C.M.; Yazawa, N.; Watanabe, R.; Ishiura, N.; Kumanogoh, A.; Okochi, H.; Tamaki, K.; Fujimoto, M.; Tedder, T.F. CD83 influences cell-surface MHC class II expression on B cells and other antigen-presenting cells. Int. Immunol. 2007, 19, 977–992. [Google Scholar] [CrossRef]
- Krzyzak, L.; Seitz, C.; Urbat, A.; Hutzler, S.; Ostalecki, C.; Glasner, J.; Hiergeist, A.; Gessner, A.; Winkler, T.H.; Steinkasserer, A.; et al. CD83 Modulates B Cell Activation and Germinal Center Responses. J. Immunol. 2016, 196, 3581–3594. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, L.F.; Appleby, M.W.; Staehling-Hampton, K.; Andrews, D.M.; Chen, Y.; McEuen, M.; Tang, P.; Rhinehart, R.L.; Proll, S.; Paeper, B.; et al. A novel mutation in CD83 results in the development of a unique population of CD4+ T cells. J. Immunol. 2004, 173, 2995–3001. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Li, S.; Shu, H.B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Jain, R.; Guan, J.; Vuong, V.; Ishido, S.; La Gruta, N.L.; Gray, D.H.; Villadangos, J.A.; Mintern, J.D. Ubiquitin ligase MARCH 8 cooperates with CD83 to control surface MHC II expression in thymic epithelium and CD4 T cell selection. J. Exp. Med. 2016, 213, 1695–1703. [Google Scholar] [CrossRef]
- Ishikawa, R.; Kajikawa, M.; Ishido, S. Loss of MHC II ubiquitination inhibits the activation and differentiation of CD4 T cells. Int. Immunol. 2014, 26, 283–289. [Google Scholar] [CrossRef]
- Oh, J.; Perry, J.S.A.; Pua, H.; Irgens-Moller, N.; Ishido, S.; Hsieh, C.S.; Shin, J.S. MARCH1 protects the lipid raft and tetraspanin web from MHCII proteotoxicity in dendritic cells. J. Cell Biol. 2018, 217, 1395–1410. [Google Scholar] [CrossRef] [Green Version]
- Ishido, S.; Kajikawa, M. MHC class II fine tuning by ubiquitination: Lesson from MARCHs. Immunogenetics 2019, 71, 197–201. [Google Scholar] [CrossRef]
- Batool, A.; Liu, H.; Liu, Y.X.; Chen, S.R. CD83, a Novel MAPK Signaling Pathway Interactor, Determines Ovarian Cancer Cell Fate. Cancers 2020, 12, 2269. [Google Scholar] [CrossRef]
- Hock, B.D.; Fernyhough, L.J.; Gough, S.M.; Steinkasserer, A.; Cox, A.G.; McKenzie, J.L. Release and clinical significance of soluble CD83 in chronic lymphocytic leukemia. Leuk. Res. 2009, 33, 1089–1095. [Google Scholar] [CrossRef]
- Lechmann, M.; Krooshoop, D.J.; Dudziak, D.; Kremmer, E.; Kuhnt, C.; Figdor, C.G.; Schuler, G.; Steinkasserer, A. The extracellular domain of CD83 inhibits dendritic cell-mediated T cell stimulation and binds to a ligand on dendritic cells. J. Exp. Med. 2001, 194, 1813–1821. [Google Scholar] [CrossRef]
- Bock, F.; Rossner, S.; Onderka, J.; Lechmann, M.; Pallotta, M.T.; Fallarino, F.; Boon, L.; Nicolette, C.; DeBenedette, M.A.; Tcherepanova, I.Y.; et al. Topical application of soluble CD83 induces IDO-mediated immune modulation, increases Foxp3+ T cells, and prolongs allogeneic corneal graft survival. J. Immunol. 2013, 191, 1965–1975. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Arp, J.; Lian, D.M.; Liu, W.H.; Baroja, M.L.; Jiang, J.F.; Ramcharran, S.; ElDeen, F.Z.; Zinser, E.; Steinkasserer, A.; et al. Immunosuppression Involving Soluble CD83 Induces Tolerogenic Dendritic Cells That Prevent Cardiac Allograft Rejection. Transplantation 2010, 90, 1145–1156. [Google Scholar] [CrossRef]
- Lan, Z.; Ge, W.; Arp, J.; Jiang, J.; Liu, W.; Gordon, D.; Healey, D.; DeBenedette, M.; Nicolette, C.; Garcia, B.; et al. Induction of kidney allograft tolerance by soluble CD83 associated with prevalence of tolerogenic dendritic cells and indoleamine 2,3-dioxygenase. Transplantation 2010, 90, 1286–1293. [Google Scholar] [CrossRef]
- Lan, Z.; Lian, D.; Liu, W.; Arp, J.; Charlton, B.; Ge, W.; Brand, S.; Healey, D.; DeBenedette, M.; Nicolette, C.; et al. Prevention of chronic renal allograft rejection by soluble CD83. Transplantation 2010, 90, 1278–1285. [Google Scholar] [CrossRef]
- Peckert-Maier, K.; Schonberg, A.; Wild, A.B.; Royzman, D.; Braun, G.; Stich, L.; Hadrian, K.; Tripal, P.; Cursiefen, C.; Steinkasserer, A.; et al. Pre-incubation of corneal donor tissue with sCD83 improves graft survival via the induction of alternatively activated macrophages and tolerogenic dendritic cells. Am. J. Transplant. 2021. [Google Scholar] [CrossRef]
- Xiong, L.; Wang, D.; Lin, S.; Wang, Y.; Luo, M.; Gao, L. Soluble CD83 inhibits acute rejection by up regulating TGF-beta and IDO secretion in rat liver transplantation. Transpl Immunol. 2021, 64, 101351. [Google Scholar] [CrossRef]
- Horvatinovich, J.M.; Grogan, E.W.; Norris, M.; Steinkasserer, A.; Lemos, H.; Mellor, A.L.; Tcherepanova, I.Y.; Nicolette, C.A.; DeBenedette, M.A. Soluble CD83 Inhibits T Cell Activation by Binding to the TLR4/MD-2 Complex on CD14(+) Monocytes. J. Immunol. 2017, 198, 2286–2301. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Cao, Z.; Wara, A.K.; Icli, B.; Sun, X.; Packard, R.R.; Esen, F.; Stapleton, C.J.; Subramaniam, M.; Kretschmer, K.; Apostolou, I.; et al. Kruppel-like factor KLF10 targets transforming growth factor-beta1 to regulate CD4(+)CD25(-) T cells and T regulatory cells. J. Biol. Chem. 2009, 284, 24914–24924. [Google Scholar] [CrossRef] [Green Version]
- Orabona, C.; Pallotta, M.T.; Grohmann, U. Different partners, opposite outcomes: A new perspective of the immunobiology of indoleamine 2,3-dioxygenase. Mol. Med. 2012, 18, 834–842. [Google Scholar] [CrossRef]
- Chen, W. IDO: More than an enzyme. Nat. Immunol. 2011, 12, 809–811. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Zhang, G.; Gao, C.; Zhong, W.; Zhang, X. CD83-stimulated monocytes suppress T-cell immune responses through production of prostaglandin E2. Proc. Natl. Acad. Sci. USA 2011, 108, 18778–18783. [Google Scholar] [CrossRef] [Green Version]
- Kotzor, N.; Lechmann, M.; Zinser, E.; Steinkasserer, A. The soluble form of CD83 dramatically changes the cytoskeleton of dendritic cells. Immunobiology 2004, 209, 129–140. [Google Scholar] [CrossRef]
- Lin, W.; Zhou, S.; Feng, M.; Yu, Y.; Su, Q.; Li, X. Soluble CD83 Regulates Dendritic Cell-T Cell Immunological Synapse Formation by Disrupting Rab1a-Mediated F-Actin Rearrangement. Front. Cell Dev. Biol. 2020, 8, 605713. [Google Scholar] [CrossRef]
- Ju, X.; Silveira, P.A.; Hsu, W.H.; Elgundi, Z.; Alingcastre, R.; Verma, N.D.; Fromm, P.D.; Hsu, J.L.; Bryant, C.; Li, Z.; et al. The Analysis of CD83 Expression on Human Immune Cells Identifies a Unique CD83+-Activated T Cell Population. J. Immunol. 2016, 197, 4613–4625. [Google Scholar] [CrossRef] [Green Version]
- Prazma, C.M.; Yazawa, N.; Fujimoto, Y.; Fujimoto, M.; Tedder, T.F. CD83 expression is a sensitive marker of activation required for B cell and CD4+ T cell longevity in vivo. J. Immunol. 2007, 179, 4550–4562. [Google Scholar] [CrossRef] [Green Version]
- Kreiser, S.; Eckhardt, J.; Kuhnt, C.; Stein, M.; Krzyzak, L.; Seitz, C.; Tucher, C.; Knippertz, I.; Becker, C.; Gunther, C.; et al. Murine CD83-positive T cells mediate suppressor functions in vitro and in vivo. Immunobiology 2015, 220, 270–279. [Google Scholar] [CrossRef]
- Chen, L.; Guan, S.; Zhou, Q.; Sheng, S.; Zhong, F.; Wang, Q. Continuous expression of CD83 on activated human CD4(+) T cells is correlated with their differentiation into induced regulatory T cells. Mol. Med. Rep. 2015, 12, 3309–3314. [Google Scholar] [CrossRef] [Green Version]
- Reinwald, S.; Wiethe, C.; Westendorf, A.M.; Breloer, M.; Probst-Kepper, M.; Fleischer, B.; Steinkasserer, A.; Buer, J.; Hansen, W. CD83 expression in CD4+ T cells modulates inflammation and autoimmunity. J. Immunol. 2008, 180, 5890–5897. [Google Scholar] [CrossRef] [Green Version]
- Zinser, E.; Naumann, R.; Wild, A.B.; Michalski, J.; Deinzer, A.; Stich, L.; Kuhnt, C.; Steinkasserer, A.; Knippertz, I. Endogenous Expression of the Human CD83 Attenuates EAE Symptoms in Humanized Transgenic Mice and Increases the Activity of Regulatory T Cells. Front. Immunol. 2019, 10, 1442. [Google Scholar] [CrossRef]
- Liedtke, K.; Alter, C.; Gunther, A.; Hovelmeyer, N.; Klopfleisch, R.; Naumann, R.; Wunderlich, F.T.; Buer, J.; Westendorf, A.M.; Hansen, W. Endogenous CD83 Expression in CD4(+) Conventional T Cells Controls Inflammatory Immune Responses. J. Immunol. 2020, 204, 3217–3226. [Google Scholar] [CrossRef]
- Bo, L.; Guojun, T.; Li, G. An Expanded Neuroimmunomodulation Axis: sCD83-Indoleamine 2,3-Dioxygenase-Kynurenine Pathway and Updates of Kynurenine Pathway in Neurologic Diseases. Front. Immunol. 2018, 9, 1363. [Google Scholar] [CrossRef]
- Wiendl, M.; Becker, E.; Muller, T.M.; Voskens, C.J.; Neurath, M.F.; Zundler, S. Targeting Immune Cell Trafficking—Insights From Research Models and Implications for Future IBD Therapy. Front. Immunol. 2021, 12, 656452. [Google Scholar] [CrossRef]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Matteoli, G.; Mazzini, E.; Iliev, I.D.; Mileti, E.; Fallarino, F.; Puccetti, P.; Chieppa, M.; Rescigno, M. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 2010, 59, 595–604. [Google Scholar] [CrossRef]
- Ciorba, M.A. Indoleamine 2,3 dioxygenase in intestinal disease. Curr. Opin. Gastroenterol. 2013, 29, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Dejban, P.; Nikravangolsefid, N.; Chamanara, M.; Dehpour, A.; Rashidian, A. The role of medicinal products in the treatment of inflammatory bowel diseases (IBD) through inhibition of TLR4/NF-kappaB pathway. Phytother. Res. 2021, 35, 835–845. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Bozec, A.; Zaiss, M.M.; Kagwiria, R.; Voll, R.; Rauh, M.; Chen, Z.; Mueller-Schmucker, S.; Kroczek, R.A.; Heinzerling, L.; Moser, M.; et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci. Transl. Med. 2014, 6, 235ra260. [Google Scholar] [CrossRef]
- Yokoyama-Kokuryo, W.; Yamazaki, H.; Takeuchi, T.; Amano, K.; Kikuchi, J.; Kondo, T.; Nakamura, S.; Sakai, R.; Hirano, F.; Nanki, T.; et al. Identification of molecules associated with response to abatacept in patients with rheumatoid arthritis. Arthritis Res. Ther. 2020, 22, 46. [Google Scholar] [CrossRef] [Green Version]
- Diehl, R.; Ferrara, F.; Muller, C.; Dreyer, A.Y.; McLeod, D.D.; Fricke, S.; Boltze, J. Immunosuppression for in vivo research: State-of-the-art protocols and experimental approaches. Cell Mol. Immunol. 2017, 14, 146–179. [Google Scholar] [CrossRef] [Green Version]
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): A harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020, 395, 1627–1639. [Google Scholar] [CrossRef]
- Xu, J.F.; Huang, B.J.; Yin, H.; Xiong, P.; Feng, W.; Xu, Y.; Fang, M.; Zheng, F.; Wang, C.Y.; Gong, F.L. A limited course of soluble CD83 delays acute cellular rejection of MHC-mismatched mouse skin allografts. Transpl. Int. 2007, 20, 266–276. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Zhuang, Q.; Peng, B.; Zhu, Y.; Ye, Q.; Ming, Y. The Evolving Roles of Macrophages in Organ Transplantation. J. Immunol. Res. 2019, 2019, 5763430. [Google Scholar] [CrossRef]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [Green Version]
- Rapp, M.; Wintergerst, M.W.M.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.L.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med. 2019, 216, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peckert-Maier, K.; Royzman, D.; Langguth, P.; Marosan, A.; Strack, A.; Sadeghi Shermeh, A.; Steinkasserer, A.; Zinser, E.; Wild, A.B. Tilting the Balance: Therapeutic Prospects of CD83 as a Checkpoint Molecule Controlling Resolution of Inflammation. Int. J. Mol. Sci. 2022, 23, 732. https://doi.org/10.3390/ijms23020732
Peckert-Maier K, Royzman D, Langguth P, Marosan A, Strack A, Sadeghi Shermeh A, Steinkasserer A, Zinser E, Wild AB. Tilting the Balance: Therapeutic Prospects of CD83 as a Checkpoint Molecule Controlling Resolution of Inflammation. International Journal of Molecular Sciences. 2022; 23(2):732. https://doi.org/10.3390/ijms23020732
Chicago/Turabian StylePeckert-Maier, Katrin, Dmytro Royzman, Pia Langguth, Anita Marosan, Astrid Strack, Atefeh Sadeghi Shermeh, Alexander Steinkasserer, Elisabeth Zinser, and Andreas B. Wild. 2022. "Tilting the Balance: Therapeutic Prospects of CD83 as a Checkpoint Molecule Controlling Resolution of Inflammation" International Journal of Molecular Sciences 23, no. 2: 732. https://doi.org/10.3390/ijms23020732
APA StylePeckert-Maier, K., Royzman, D., Langguth, P., Marosan, A., Strack, A., Sadeghi Shermeh, A., Steinkasserer, A., Zinser, E., & Wild, A. B. (2022). Tilting the Balance: Therapeutic Prospects of CD83 as a Checkpoint Molecule Controlling Resolution of Inflammation. International Journal of Molecular Sciences, 23(2), 732. https://doi.org/10.3390/ijms23020732