
Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells

 , , , , ,
, , , , , 
Abstract
:
1. Introduction
2. Results
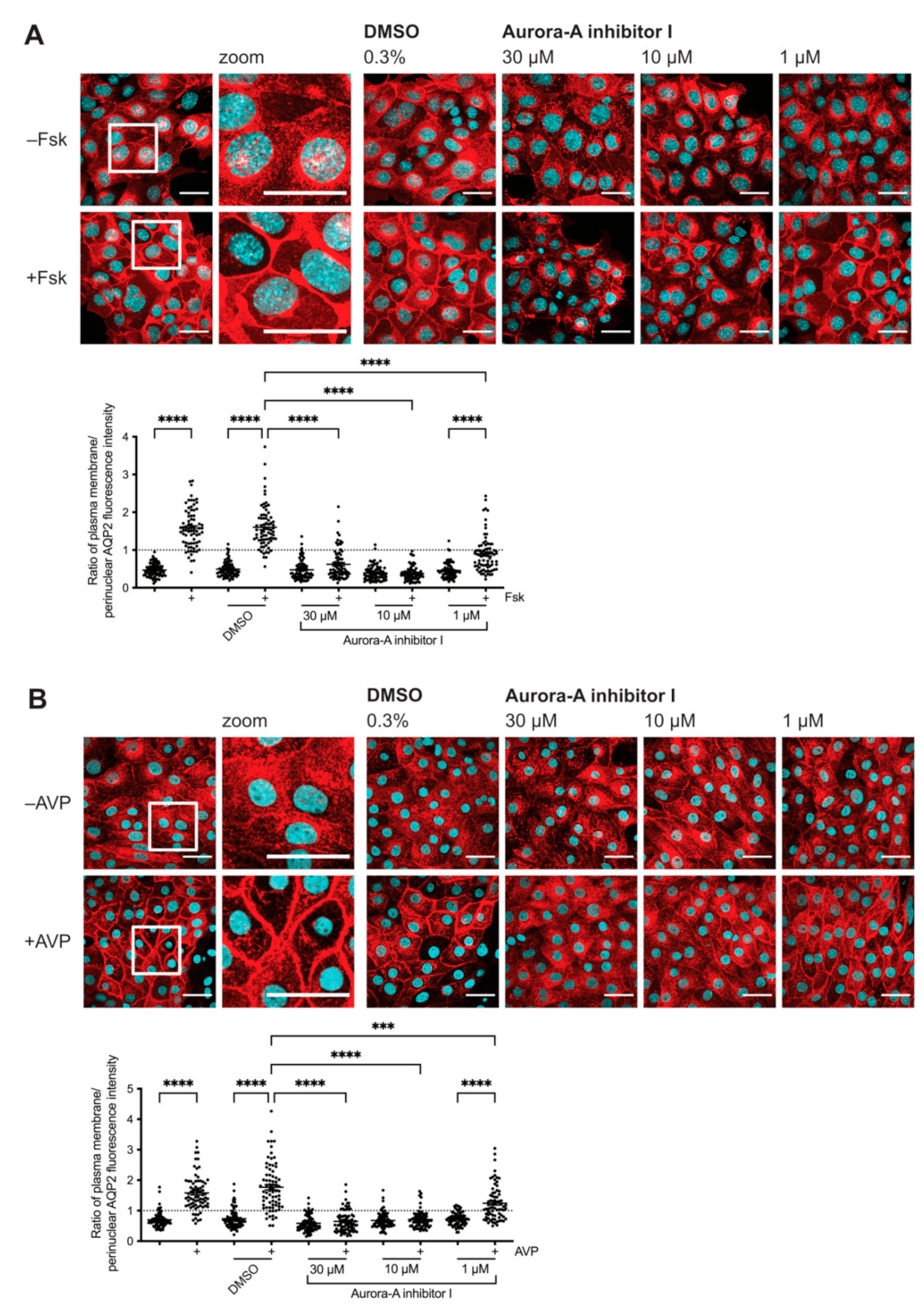
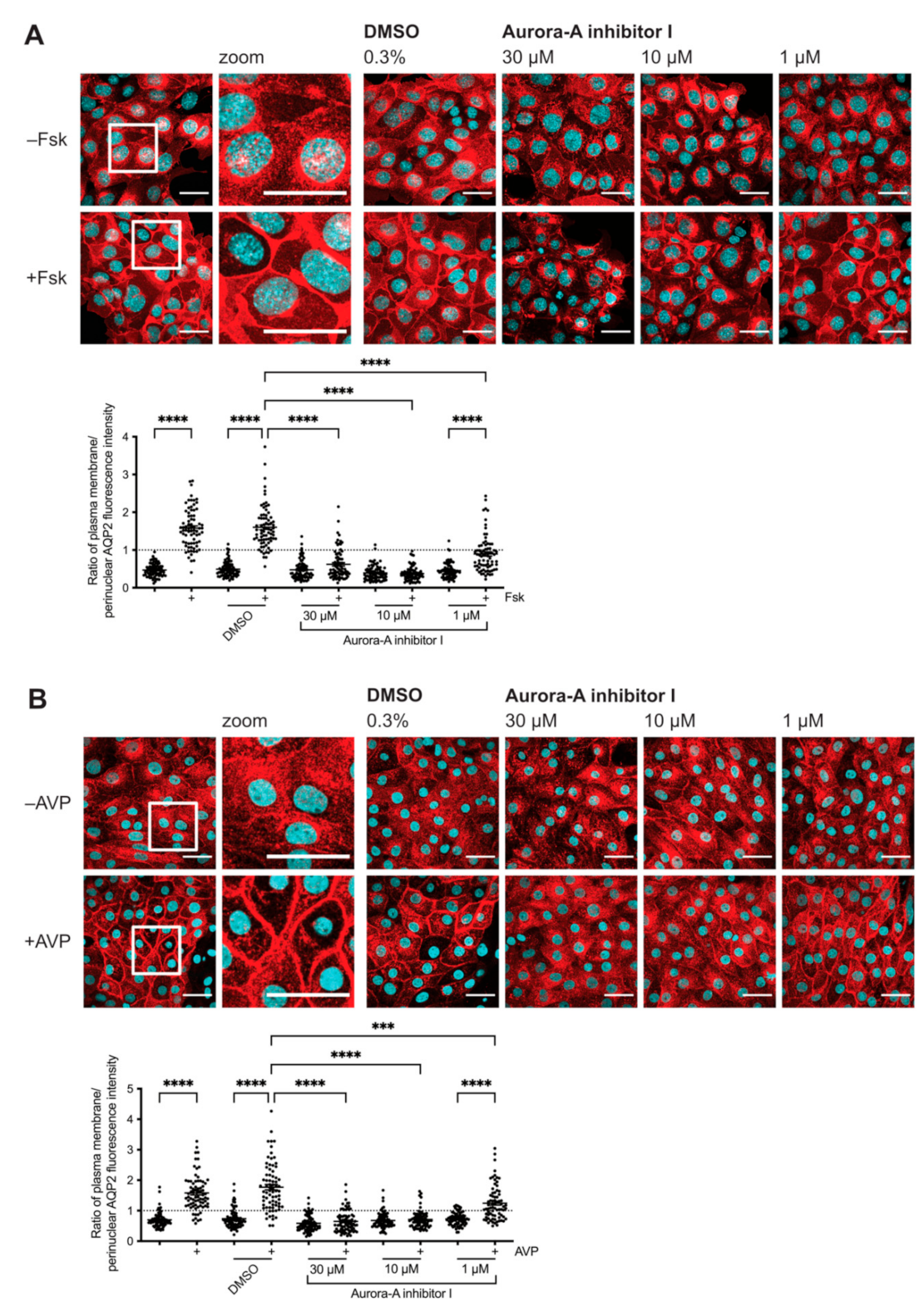
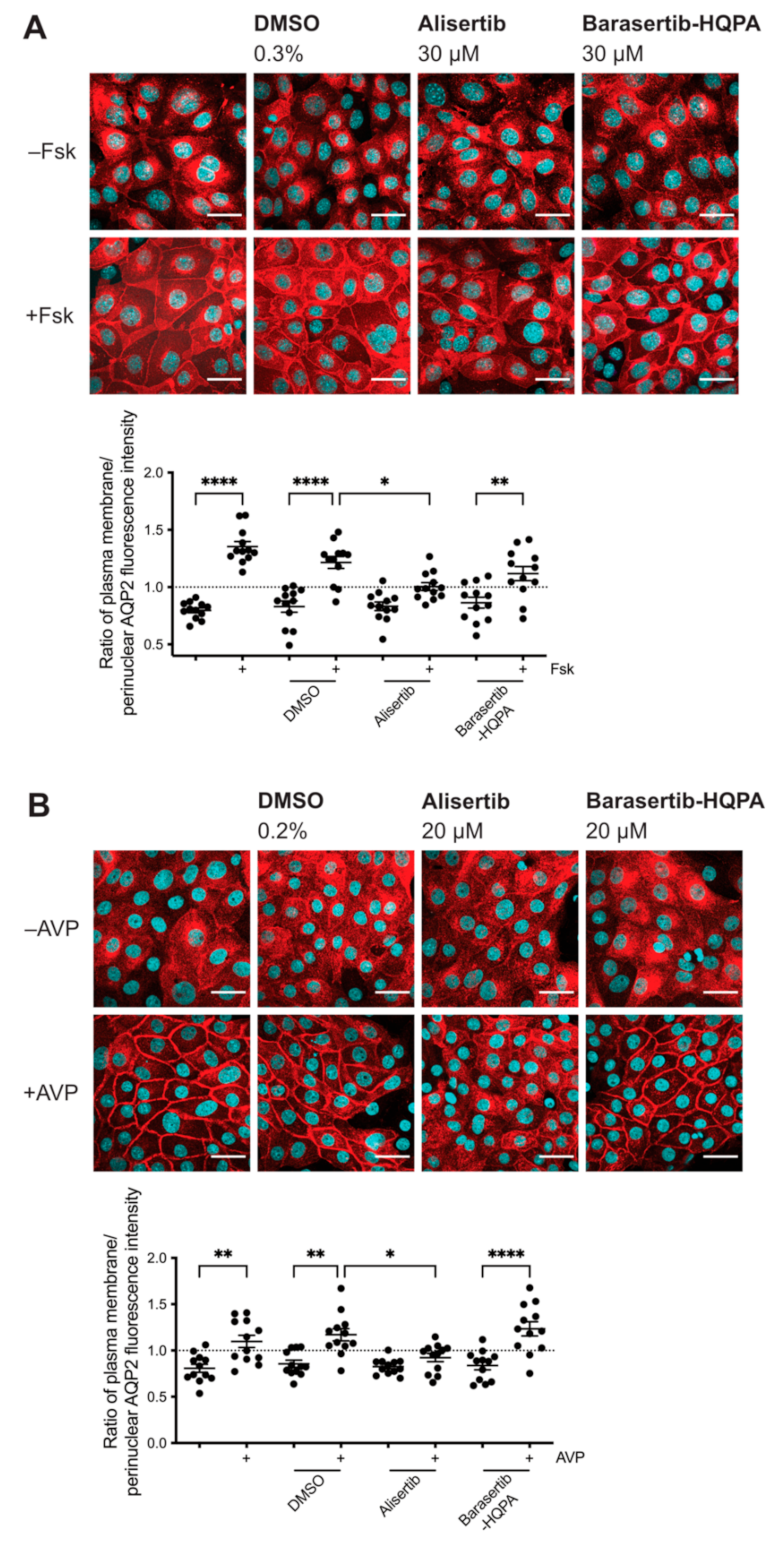
2.1. Inhibition of AURKA Prevents the cAMP-Induced Redistribution of AQP2
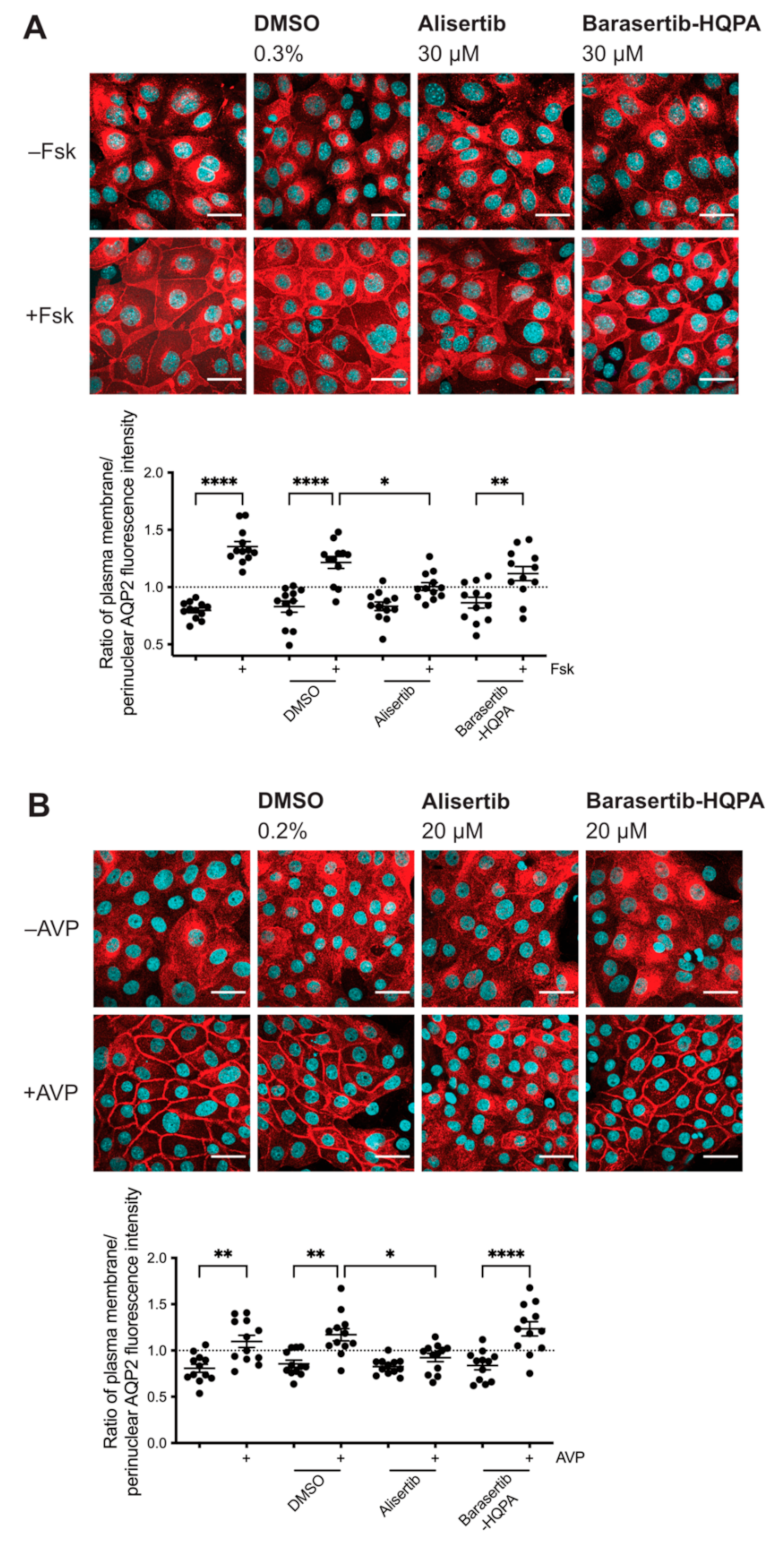
2.2. Inhibition of AURKA but Not AURKB Prevents the cAMP-Induced Redistribution of AQP2
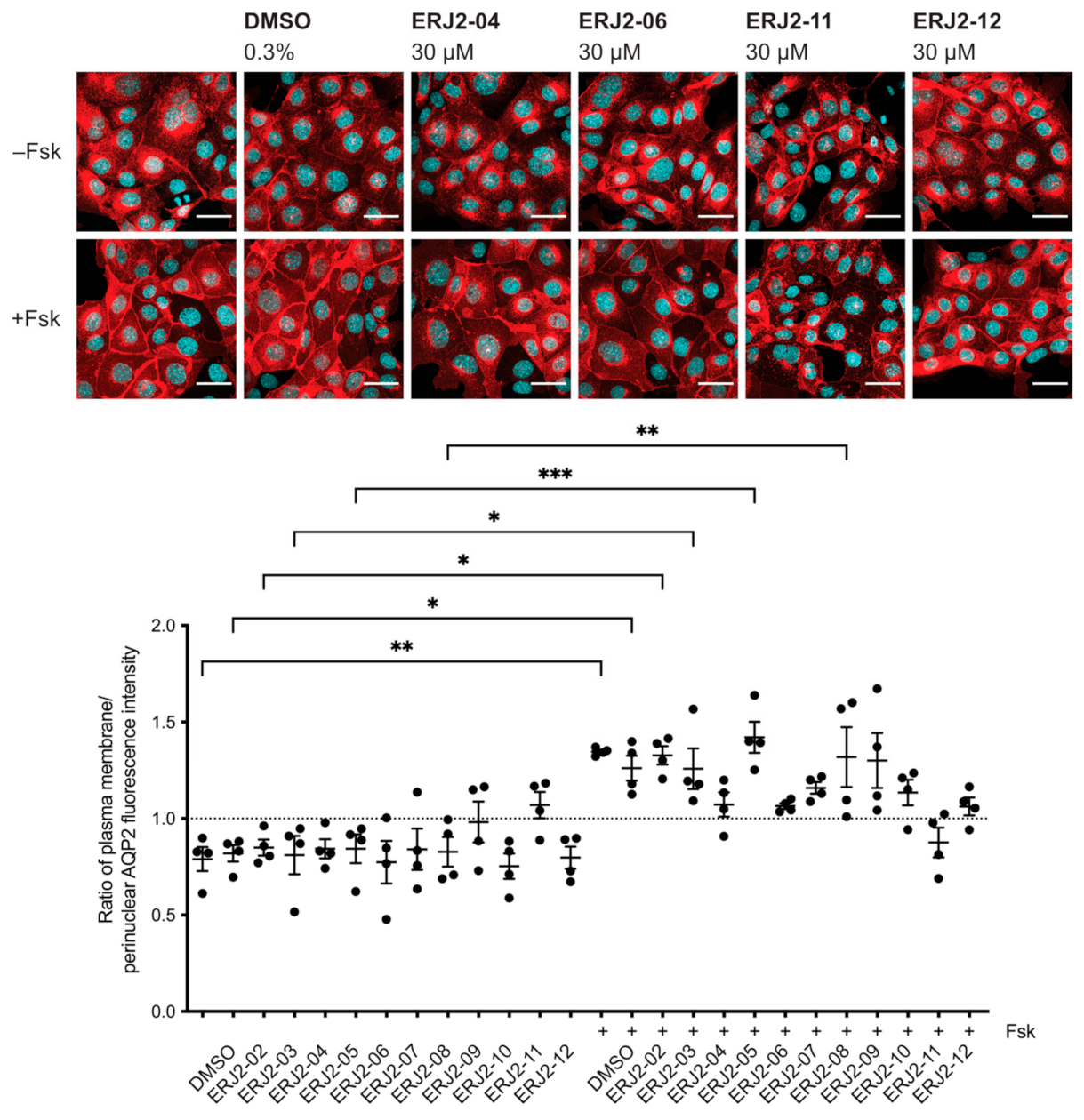
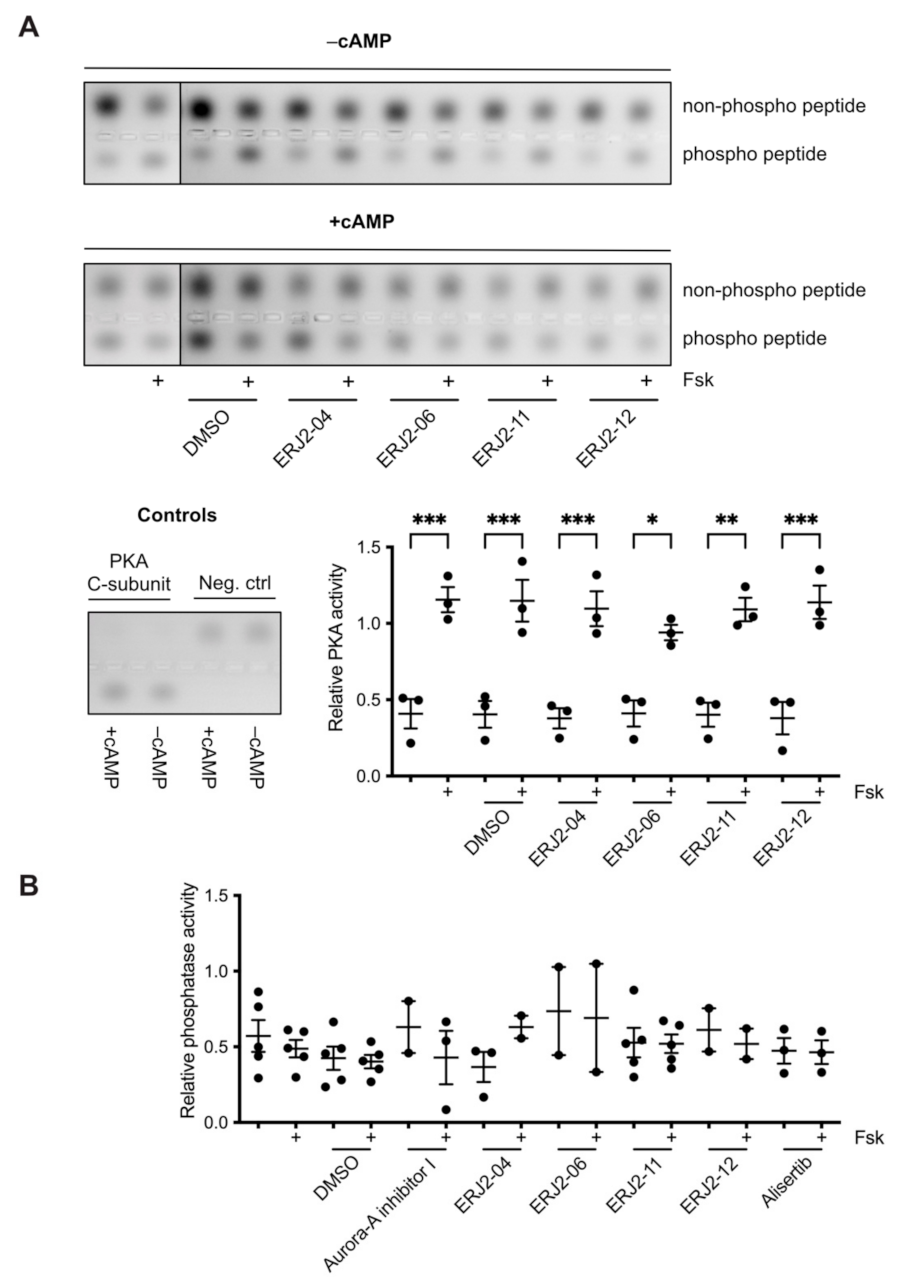
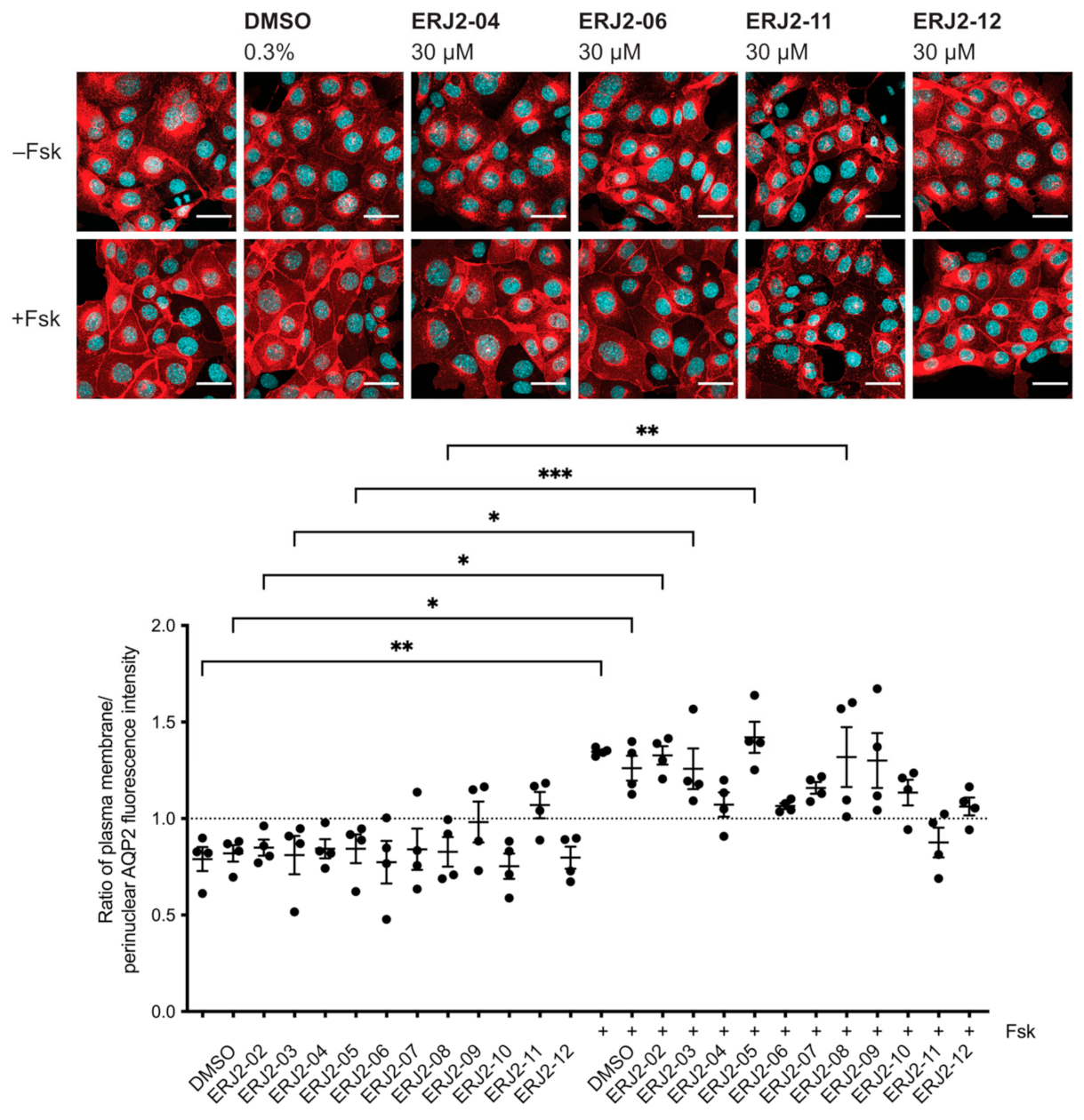
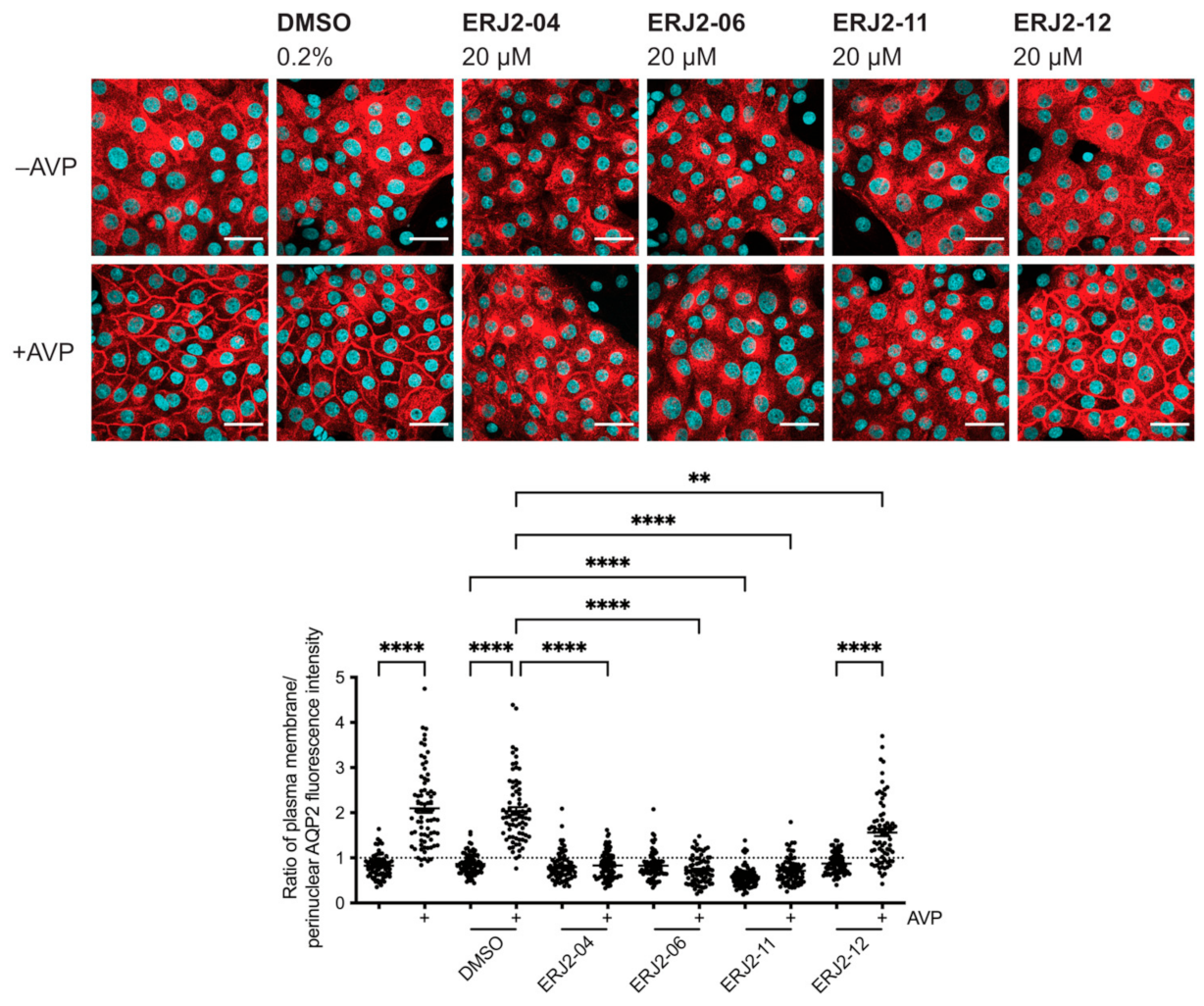
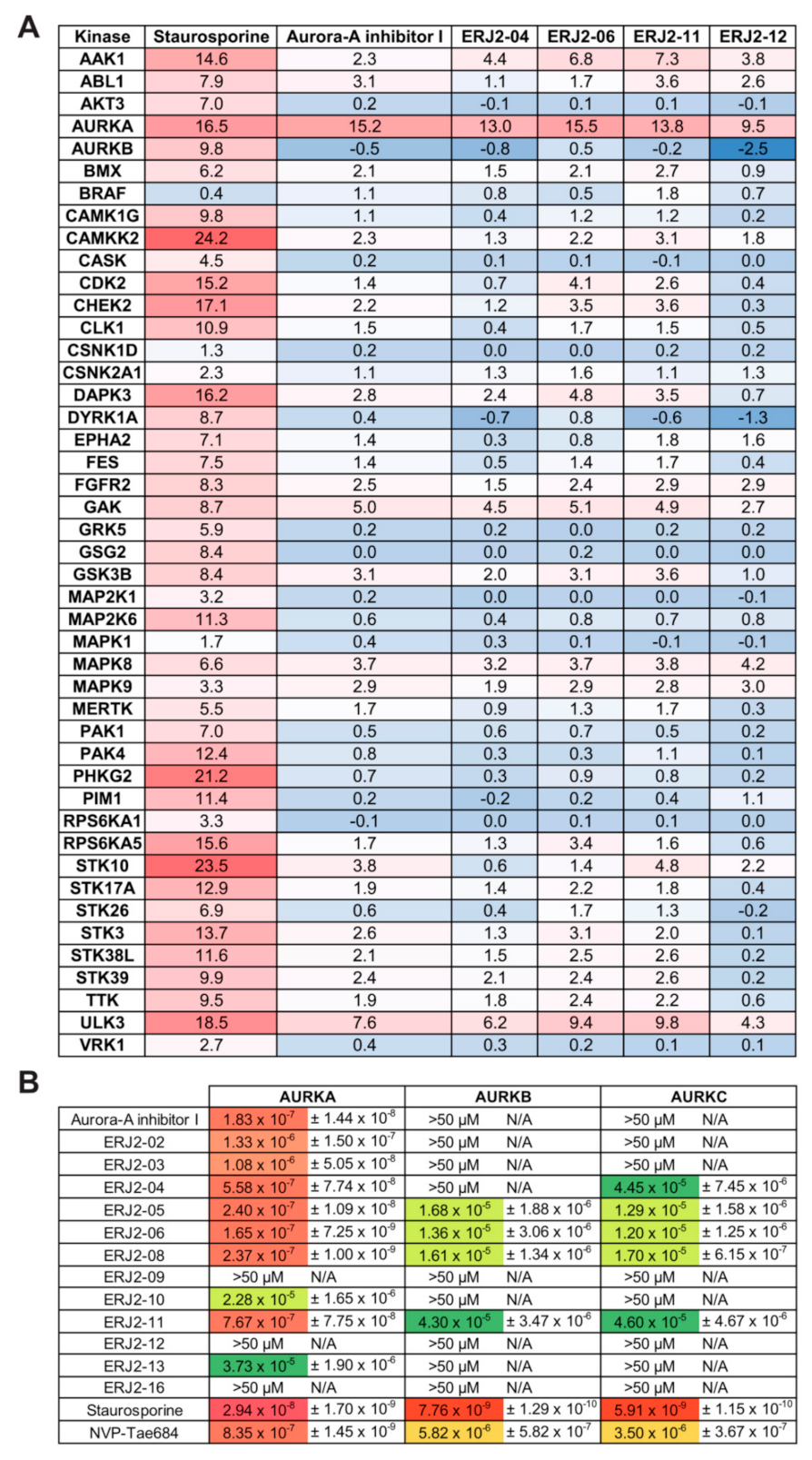
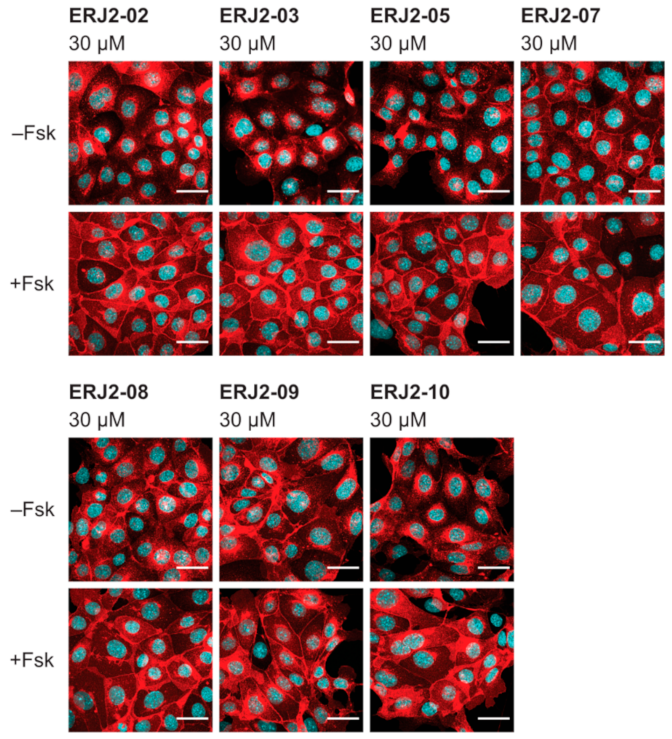
2.3. Aurora-A Inhibitor I Derivatives Show Varying Effects on the AQP2 Localization
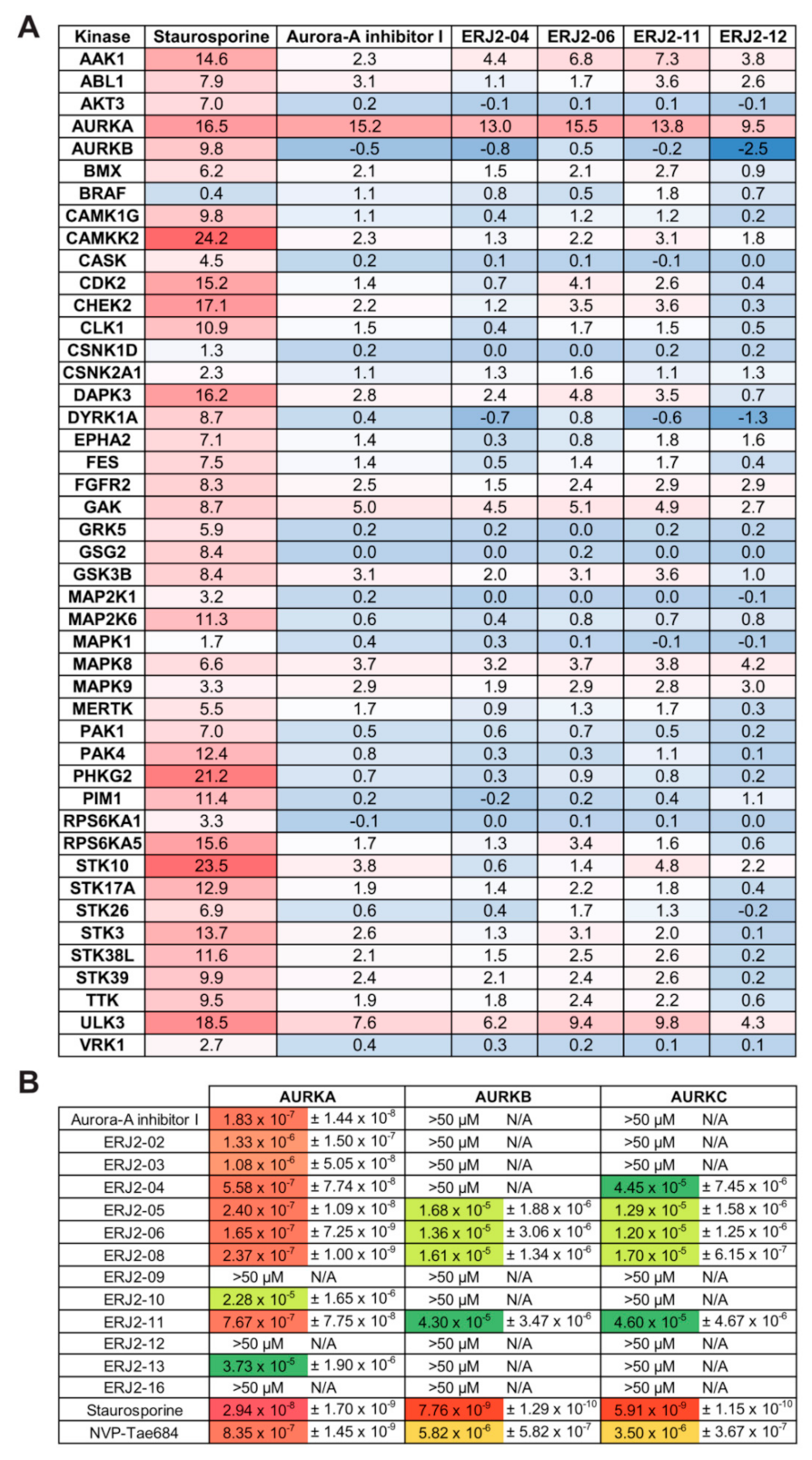
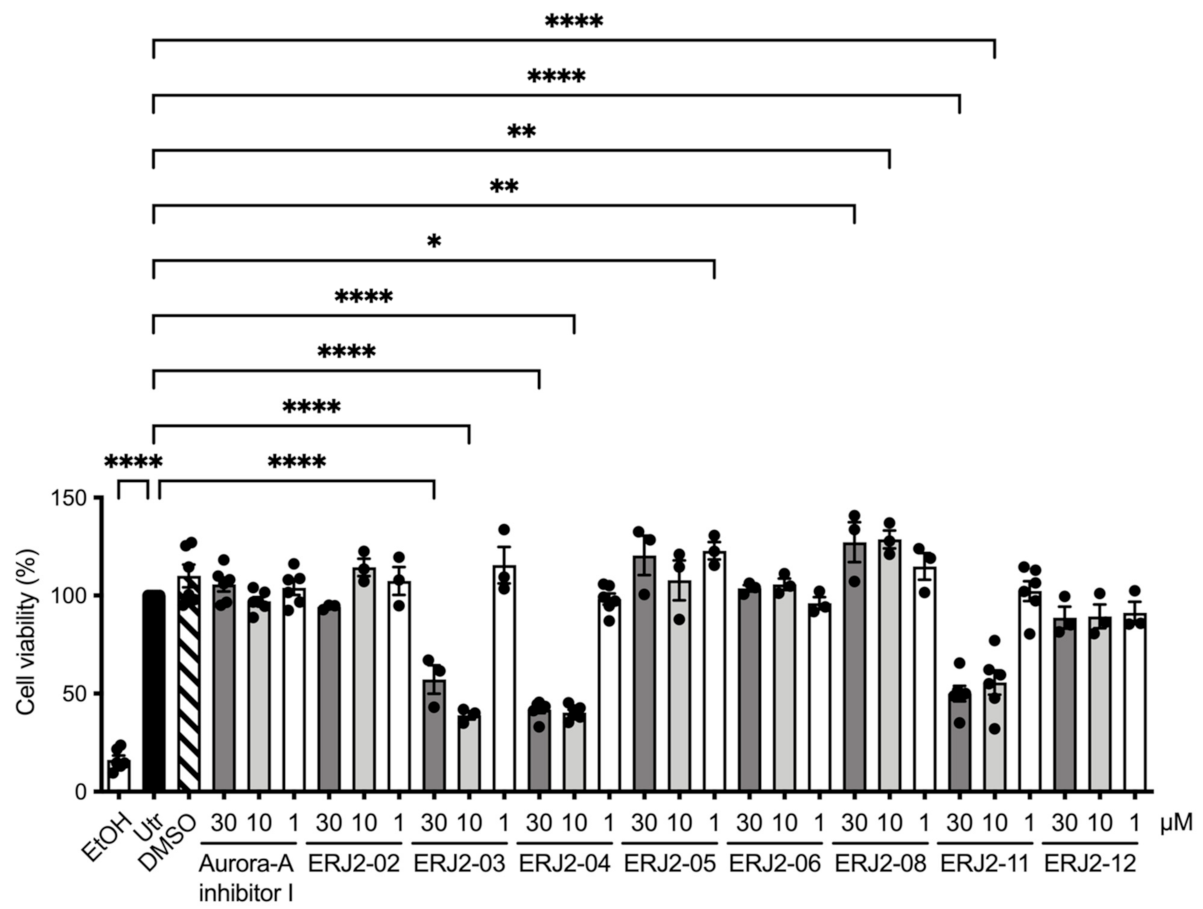
2.4. Cell Viability Is Affected by Some of the Aurora-A Inhibitor I Derivatives
2.5. Aurora-A Inhibitor I and Its Derivatives Show Favorable Selectivity Profiles
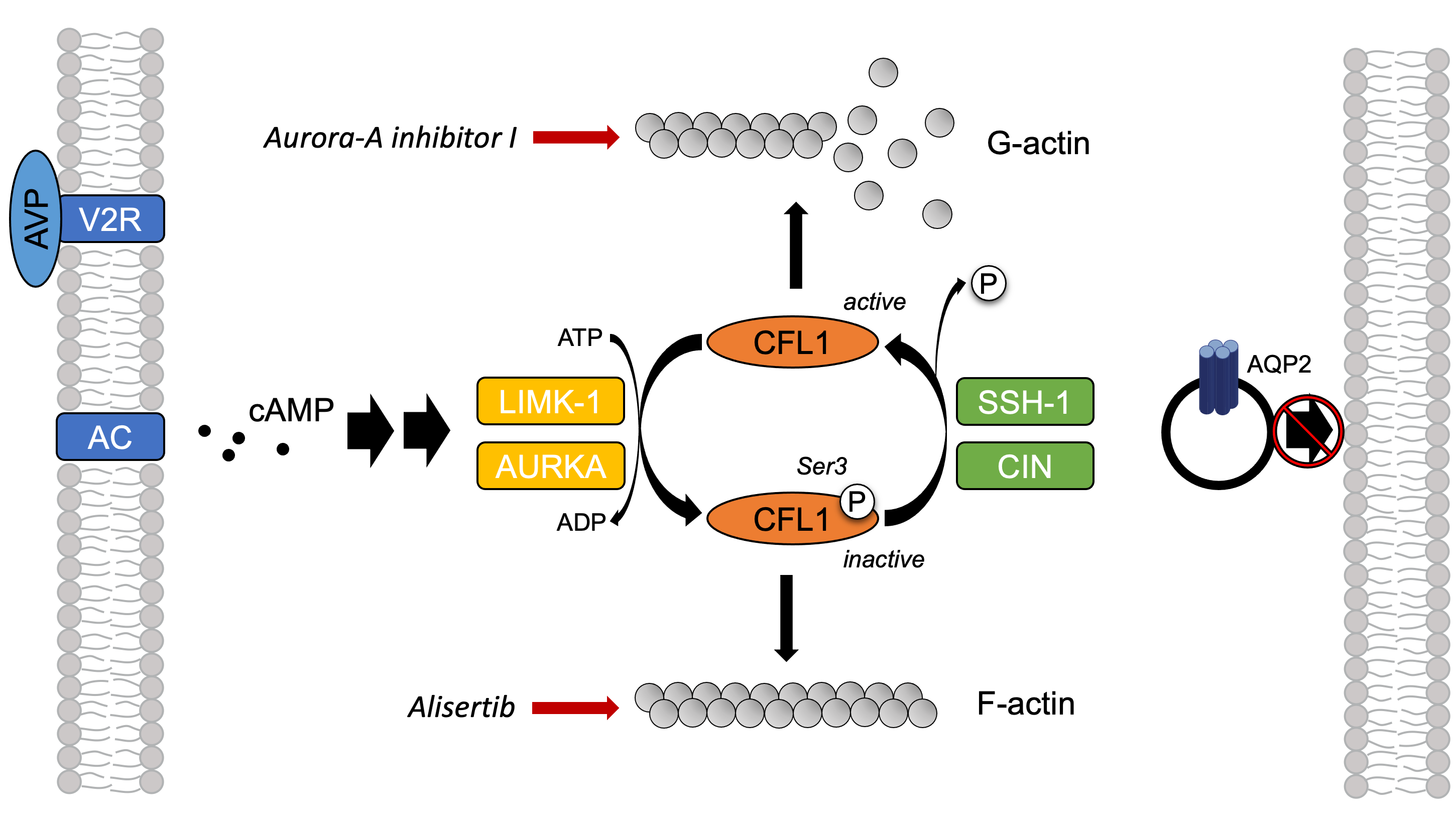
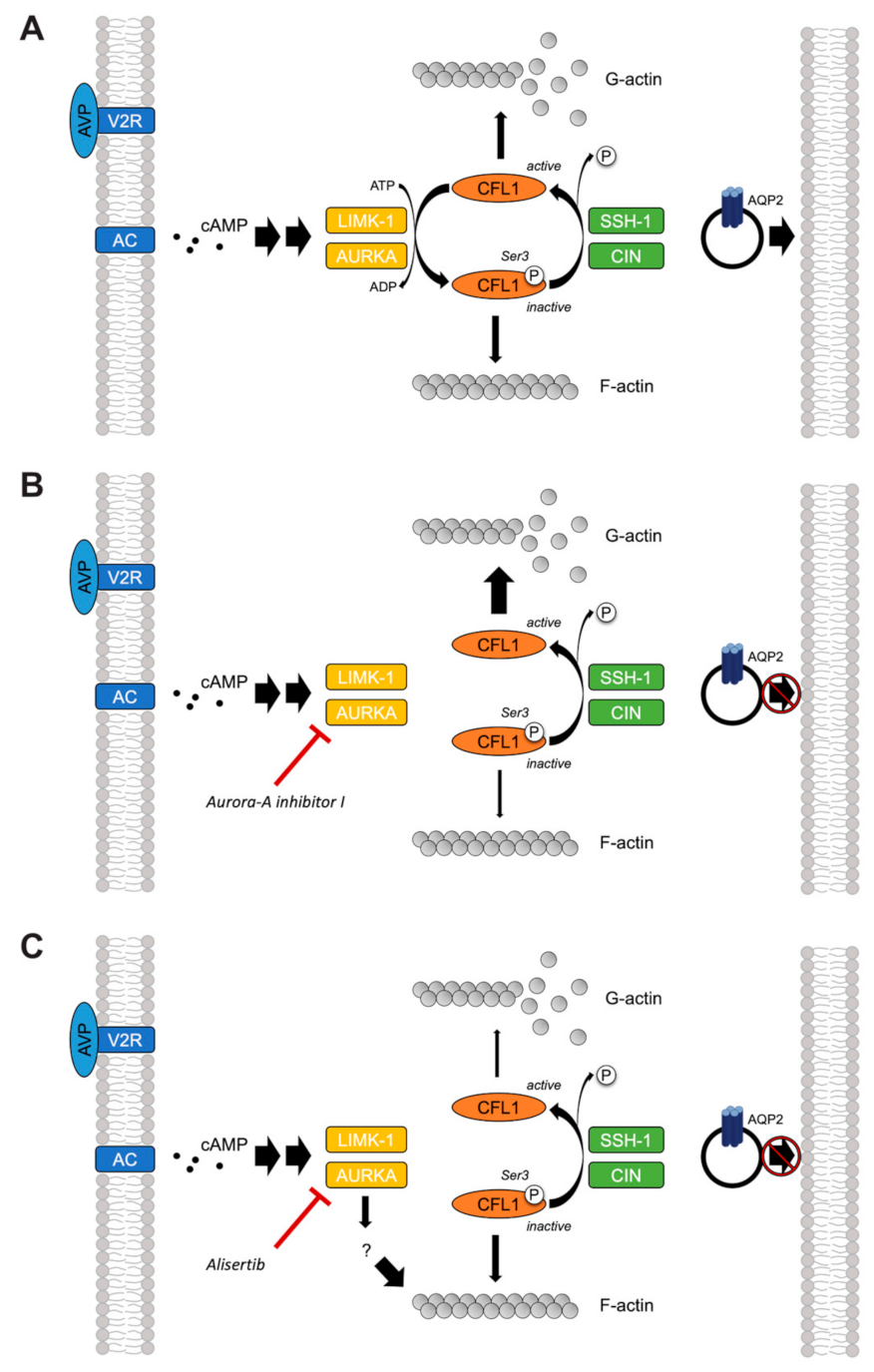
2.6. The Inhibition of AURKA Affects the Organization of the Actin Cytoskeleton in Both MCD4 and Primary IMCD Cells
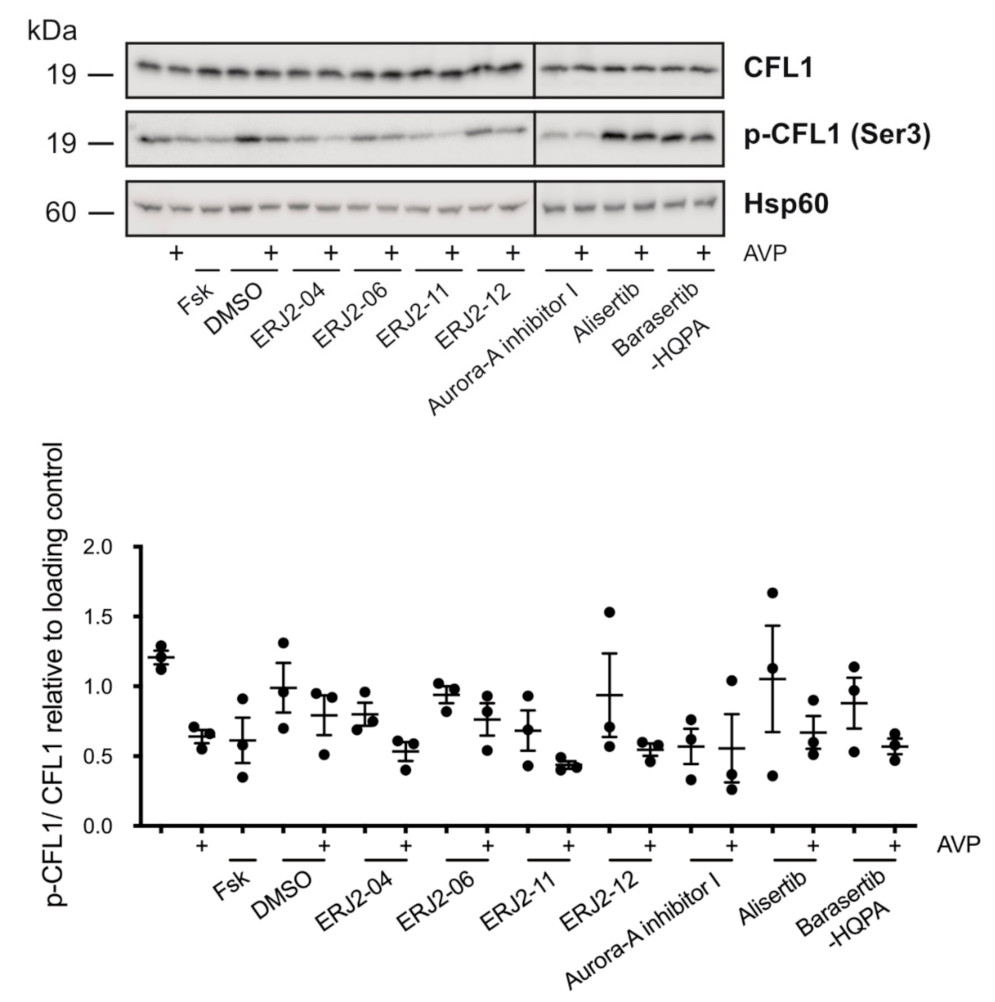
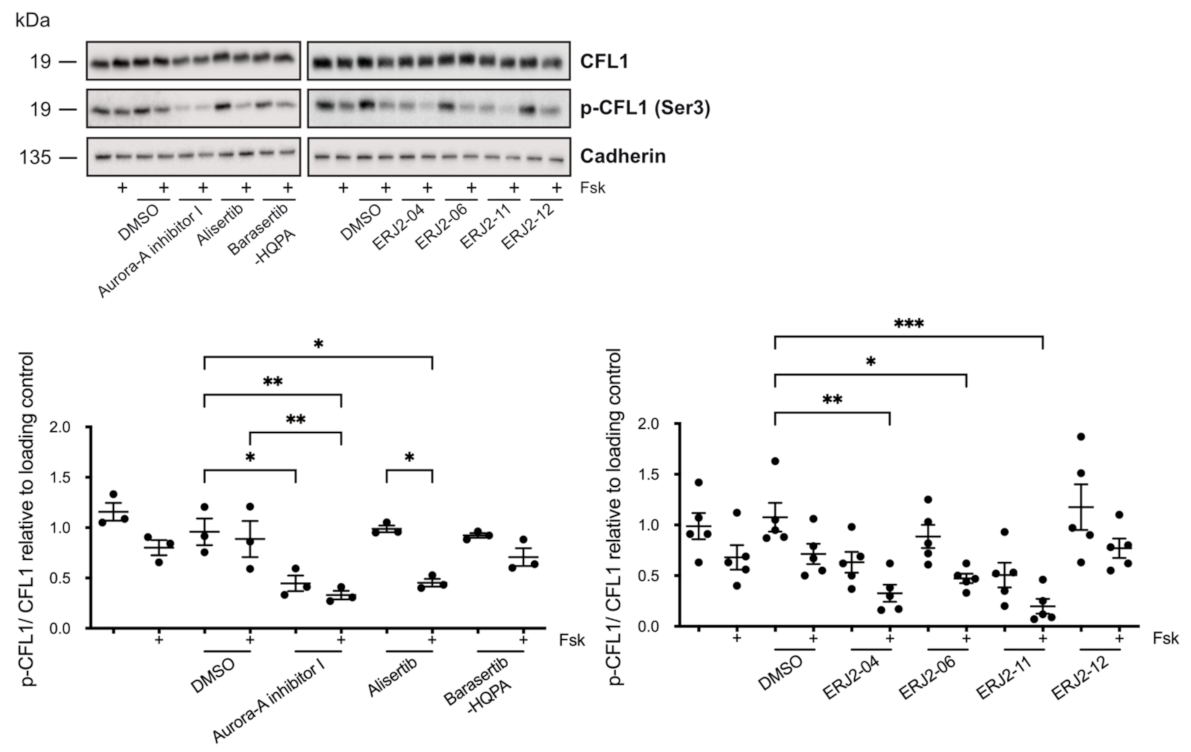
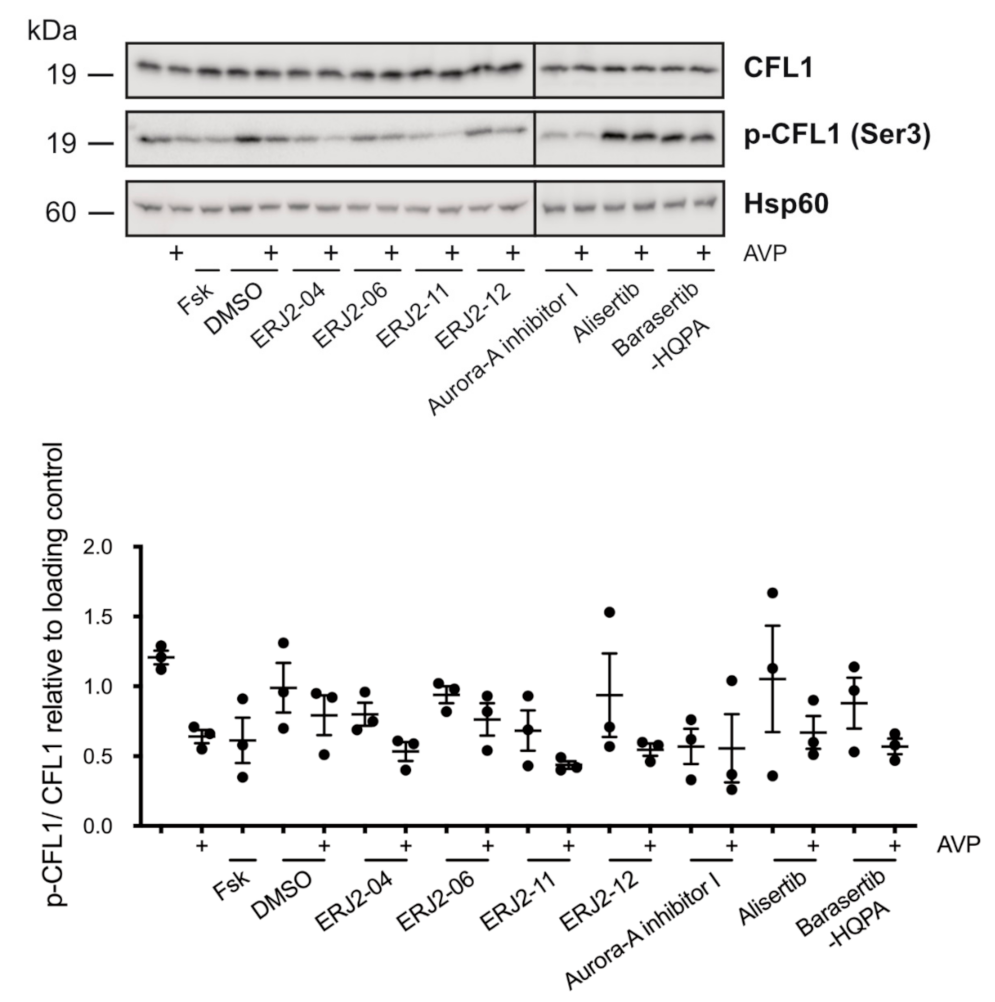
2.7. The Inhibition of AURKA by Aurora-A Inhibitor I Decreases the Phosphorylation of the AURKA Substrate CFL1 in Both MCD4 and Primary IMCD Cells
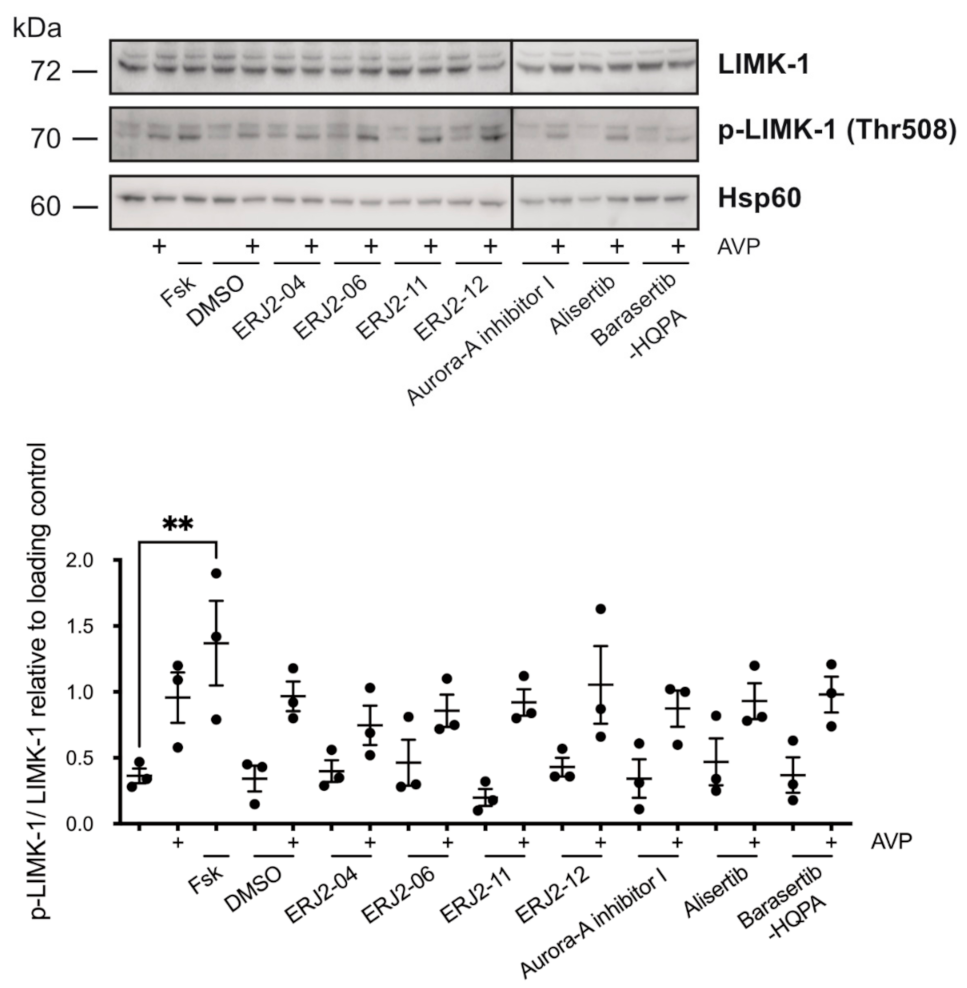
2.8. The Inhibition of AURKA and AURKB Has no Effect on the Phosphorylation of LIM Kinase-1 (LIMK-1) in Primary IMCD Cells
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis of Novel Aurora-A Inhibitor I Derivatives
4.2. Compounds
4.3. Cell Cultures
4.4. Animal Treatment
4.5. Immunofluorescence Microscopy of MCD4 and Primary IMCD Cells
4.6. MTS Assays
4.7. Western Blotting
4.8. Differential Scanning Fluorimetry-Based Selectivity Screening against a Curated Kinase Library (Thermal Shift Assay)
4.9. NanoBRET Assay
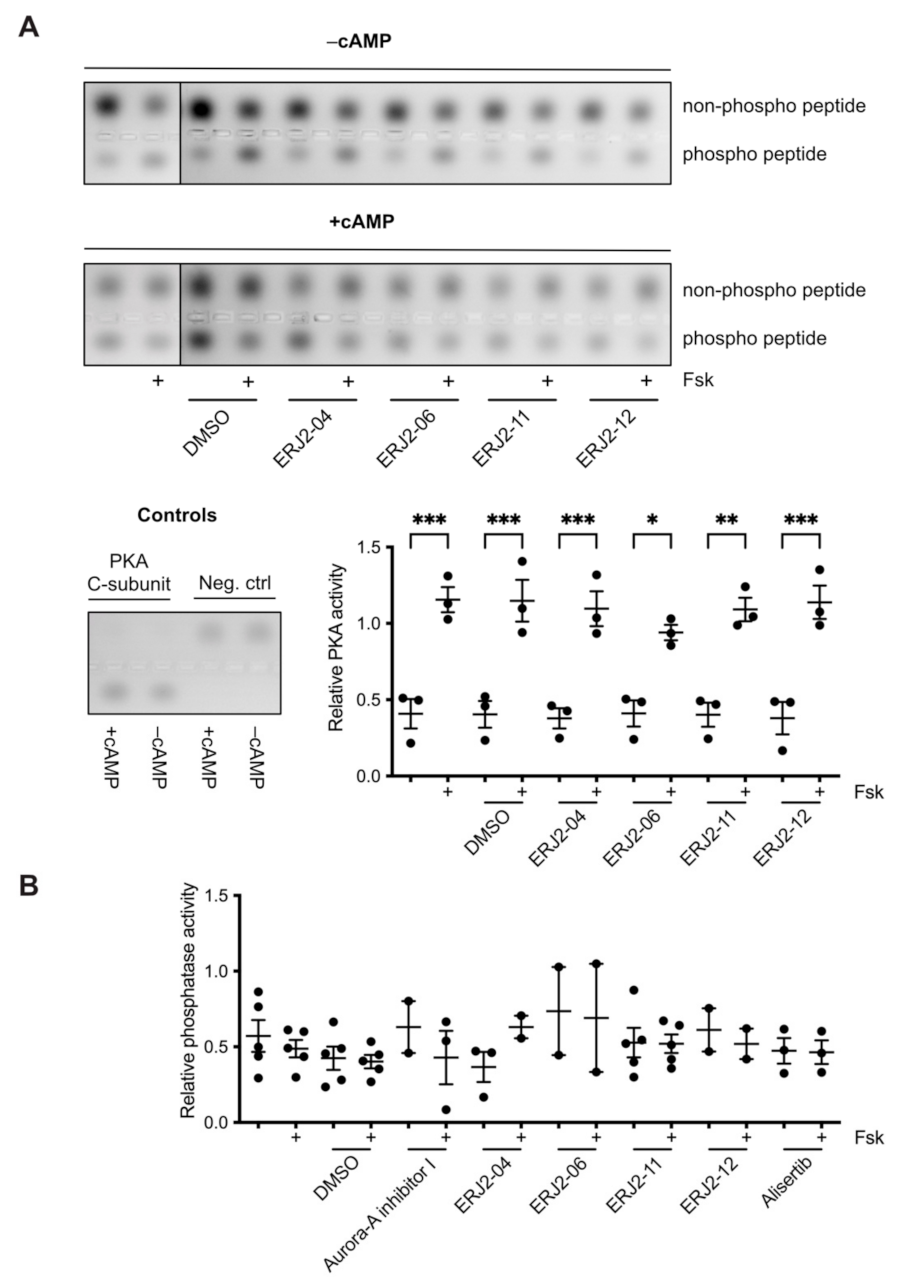
4.10. PKA Activity Assay
4.11. Phosphatase Activity Assay
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Baltzer, S.; Klussmann, E. Small molecules for modulating the localisation of the water channel aquaporin-2-disease relevance and perspectives for targeting local cAMP signalling. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Vukicevic, T.; Schulz, M.; Faust, D.; Klussmann, E. The Trafficking of the Water Channel Aquaporin-2 in Renal Principal Cells-a Potential Target for Pharmacological Intervention in Cardiovascular Diseases. Front. Pharmacol. 2016, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Olesen, E.T.B.; Fenton, R.A. Aquaporin 2 regulation: Implications for water balance and polycystic kidney diseases. Nat. Rev. Nephrol. 2021, 17, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E.; Tamma, G.; Lorenz, D.; Wiesner, B.; Maric, K.; Hofmann, F.; Aktories, K.; Valenti, G.; Rosenthal, W. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 2001, 276, 20451–20457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamma, G.; Klussmann, E.; Maric, K.; Aktories, K.; Svelto, M.; Rosenthal, W.; Valenti, G. Rho inhibits cAMP-induced translocation of aquaporin-2 into the apical membrane of renal cells. Am. J. Physiol. Renal. Physiol. 2001, 281, F1092–F1101. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.; Gao, Y.; Franki, N.; Hays, R.M. Vasopressin depolymerizes apical F-actin in rat inner medullary collecting duct. Am. J. Physiol. 1993, 265, C757–C762. [Google Scholar] [CrossRef] [PubMed]
- Nedvetsky, P.I.; Stefan, E.; Frische, S.; Santamaria, K.; Wiesner, B.; Valenti, G.; Hammer, J.A.; Nielsen, S.; Goldenring, J.R.; Rosenthal, W.; et al. A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic 2007, 8, 110–123. [Google Scholar] [CrossRef]
- Sasaki, S.; Yui, N.; Noda, Y. Actin directly interacts with different membrane channel proteins and influences channel activities: AQP2 as a model. Biochim. Et Biophys. Acta (BBA) Biomembr. 2014, 1838, 514–520. [Google Scholar] [CrossRef] [Green Version]
- Eitzen, G. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 2003, 1641, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Loo, C.S.; Chen, C.W.; Wang, P.J.; Chen, P.Y.; Lin, S.Y.; Khoo, K.H.; Fenton, R.A.; Knepper, M.A.; Yu, M.J. Quantitative apical membrane proteomics reveals vasopressin-induced actin dynamics in collecting duct cells. Proc. Natl. Acad. Sci. USA 2013, 110, 17119–17124. [Google Scholar] [CrossRef] [Green Version]
- Miklavc, P.; Frick, M. Actin and Myosin in Non-Neuronal Exocytosis. Cells 2020, 9, 1455. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Horikawa, S.; Katayama, Y.; Sasaki, S. Water channel aquaporin-2 directly binds to actin. Biochem. Biophys. Res. Commun. 2004, 322, 740–745. [Google Scholar] [CrossRef]
- Noda, Y.; Sasaki, S. Regulation of aquaporin-2 trafficking and its binding protein complex. Biochim. Et Biophys. Acta (BBA) Biomembr. 2006, 1758, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajika, Y.; Matsuzaki, T.; Suzuki, T.; Ablimit, A.; Aoki, T.; Hagiwara, H.; Kuwahara, M.; Sasaki, S.; Takata, K. Differential regulation of AQP2 trafficking in endosomes by microtubules and actin filaments. Histochem. Cell Biol. 2005, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Pisitkun, T.; Yu, M.J.; Chou, C.L.; Verbalis, M.J.; Shen, R.F.; Knepper, M.A. Large scale protein identification in intracellular aquaporin-2 vesicles from renal inner medullary collecting duct. Mol. Cell. Proteomics 2005, 4, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Noda, Y.; Horikawa, S.; Kanda, E.; Yamashita, M.; Meng, H.; Eto, K.; Li, Y.; Kuwahara, M.; Hirai, K.; Pack, C.; et al. Reciprocal interaction with G-actin and tropomyosin is essential for aquaporin-2 trafficking. J. Cell Biol. 2008, 182, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Carmena, M.; Earnshaw, W.C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Bolanos-Garcia, V.M. Aurora kinases. Int. J. Biochem. Cell Biol. 2005, 37, 1572–1577. [Google Scholar] [CrossRef]
- Fu, J.; Bian, M.; Liu, J.; Jiang, Q.; Zhang, C. A single amino acid change converts Aurora-A into Aurora-B-like kinase in terms of partner specificity and cellular function. Proc. Natl. Acad. Sci. USA 2009, 106, 6939–6944. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Deng, Z.; Fu, J.; Xu, C.; Xin, G.; Wu, Z.; Luo, J.; Wang, G.; Zhang, S.; Zhang, B.; et al. Spatial Compartmentalization Specializes the Function of Aurora A and Aurora B. J. Biol. Chem. 2015, 290, 17546–17558. [Google Scholar] [CrossRef] [Green Version]
- Hans, F.; Skoufias, D.A.; Dimitrov, S.; Margolis, R.L. Molecular distinctions between Aurora A and B: A single residue change transforms Aurora A into correctly localized and functional Aurora B. Mol. Biol. Cell 2009, 20, 3491–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, J.R.; Anderson, L.; Zhu, Y.; Mossie, K.; Ng, L.; Souza, B.; Schryver, B.; Flanagan, P.; Clairvoyant, F.; Ginther, C.; et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998, 17, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S. The potential role of Aurora kinase inhibitors in haematological malignancies. Br. J. Haematol. 2011, 155, 561–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsuka, M.; Katayama, H.; Ota, T.; Tanaka, T.; Odashima, S.; Suzuki, F.; Terada, Y. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998, 58, 4811–4816. [Google Scholar]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef]
- Katayama, H.; Sen, S. Aurora kinase inhibitors as anticancer molecules. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2010, 1799, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Aliagas-Martin, I.; Burdick, D.; Corson, L.; Dotson, J.; Drummond, J.; Fields, C.; Huang, O.W.; Hunsaker, T.; Kleinheinz, T.; Krueger, E.; et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J. Med. Chem. 2009, 52, 3300–3307. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Adhikari, B.; Bozilovic, J.; Diebold, M.; Schwarz, J.D.; Hofstetter, J.; Schroder, M.; Wanior, M.; Narain, A.; Vogt, M.; Stankovic, N.D.; et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 2020, 16, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ascanelli, C.; Abdelbaki, A.; Fung, A.; Rasmusson, T.; Michaelides, I.; Roberts, K.; Lindon, C. Selective targeting of non-centrosomal AURKA functions through use of a targeted protein degradation tool. Commun. Biol. 2021, 4, 640. [Google Scholar] [CrossRef]
- Ritchey, L.; Chakrabarti, R. Aurora A kinase modulates actin cytoskeleton through phosphorylation of Cofilin: Implication in the mitotic process. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2719–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dema, A.; Faust, D.; Lazarow, K.; Wippich, M.; Neuenschwander, M.; Zuhlke, K.; Geelhaar, A.; Pallien, T.; Hallscheidt, E.; Eichhorst, J.; et al. Cyclin-Dependent Kinase 18 Controls Trafficking of Aquaporin-2 and Its Abundance through Ubiquitin Ligase STUB1, Which Functions as an AKAP. Cells 2020, 9, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Lyu, J.; Yang, E.J.; Liu, Y.; Zhang, B.; Shim, J.S. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat. Commun. 2018, 9, 3212. [Google Scholar] [CrossRef]
- Vukicevic, T.; Hinze, C.; Baltzer, S.; Himmerkus, N.; Quintanova, C.; Zuhlke, K.; Compton, F.; Ahlborn, R.; Dema, A.; Eichhorst, J.; et al. Fluconazole Increases Osmotic Water Transport in Renal Collecting Duct through Effects on Aquaporin-2 Trafficking. J. Am. Soc. Nephrol. 2019, 30, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Schrade, K.; Troger, J.; Eldahshan, A.; Zuhlke, K.; Abdul Azeez, K.R.; Elkins, J.M.; Neuenschwander, M.; Oder, A.; Elkewedi, M.; Jaksch, S.; et al. An AKAP-Lbc-RhoA interaction inhibitor promotes the translocation of aquaporin-2 to the plasma membrane of renal collecting duct principal cells. PLoS ONE 2018, 13, e0191423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, M.G.; Ecsedy, J.A.; Chakravarty, A.; Silverman, L.; Zhang, M.; Hoar, K.M.; Stroud, S.G.; Chen, W.; Shinde, V.; Huck, J.J.; et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 2011, 17, 7614–7624. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.; Calabrese, E.; Hideshima, T.; Ecsedy, J.; Perrone, G.; Mani, M.; Ikeda, H.; Bianchi, G.; Hu, Y.; Cirstea, D.; et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 2010, 115, 5202–5213. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, R.W.; Odedra, R.; Heaton, S.P.; Wedge, S.R.; Keen, N.J.; Crafter, C.; Foster, J.R.; Brady, M.C.; Bigley, A.; Brown, E.; et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res. 2007, 13, 3682–3688. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P.; et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 2007, 110, 2034–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmied, C.; Baltzer, S.; Lehmann, M.; Klussmann, E. A segmentation-based approach for the quantification of aquaporin-2 (AQP2) located at the plasma membrane and perinuclear area of renal collecting duct cells. Zenodo 2021. [Google Scholar] [CrossRef]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Owen, T.C.; Cory, A.H.; Cory, J.G. 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl) tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorg. Med. Chem. Lett. 1991, 1, 611–614. [Google Scholar]
- Tzara, A.; Xanthopoulos, D.; Kourounakis, A.P. Morpholine As a Scaffold in Medicinal Chemistry: An Update on Synthetic Strategies. Chem. Med. Chem. 2020, 15, 392–403. [Google Scholar] [CrossRef]
- Kourounakis, A.P.; Xanthopoulos, D.; Tzara, A. Morpholine as a privileged structure: A review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev. 2020, 40, 709–752. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. In Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2011; Volume 795, pp. 109–118. [Google Scholar]
- Datta, A.; Yang, C.R.; Salhadar, K.; Park, E.; Chou, C.L.; Raghuram, V.; Knepper, M.A. Phosphoproteomic identification of vasopressin-regulated protein kinases in collecting duct cells. Br. J. Pharmacol. 2021, 178, 1426–1444. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Yu, M.J.; Wang, G.; Boja, E.S.; Hoffert, J.D.; Pisitkun, T.; Knepper, M.A. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3882–3887. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.X.; Van Hoek, A.; Huang, Y.; Bouley, R.; McLaughlin, M.; Brown, D. Aquaporin-2 localization in clathrin-coated pits: Inhibition of endocytosis by dominant-negative dynamin. Am. J. Physiol. Renal. Physiol. 2002, 282, F998–F1011. [Google Scholar] [CrossRef] [Green Version]
- Uawithya, P.; Pisitkun, T.; Ruttenberg, B.E.; Knepper, M.A. Transcriptional profiling of native inner medullary collecting duct cells from rat kidney. Physiol. Genom. 2008, 32, 229–253. [Google Scholar] [CrossRef] [Green Version]
- Conner, S.D.; Schmid, S.L. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol. 2002, 156, 921–929. [Google Scholar] [CrossRef]
- Zhang, C.X.; Engqvist-Goldstein, A.E.; Carreno, S.; Owen, D.J.; Smythe, E.; Drubin, D.G. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic 2005, 6, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nagamori, I.; Yabuta, N.; Nojima, H. GAK, a regulator of clathrin-mediated membrane traffic, also controls centrosome integrity and chromosome congression. J. Cell Sci. 2009, 122, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Chaikuad, A.; Keates, T.; Vincke, C.; Kaufholz, M.; Zenn, M.; Zimmermann, B.; Gutiérrez, C.; Zhang, R.G.; Hatzos-Skintges, C.; Joachimiak, A.; et al. Structure of cyclin G-associated kinase (GAK) trapped in different conformations using nanobodies. Biochem. J. 2014, 459, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Mathea, S.; Salah, E.; Tallant, C.; Chatterjee, D.; Berger, B.T.; Konietzny, R.; Müller, S.; Kessler, B.M.; Knapp, S. Conformational plasticity of the ULK3 kinase domain. Biochem. J. 2021, 478, 2811–2823. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Kasak, L.; Peil, L.; Østerlund, T.; Kogerman, P. Dual function of UNC-51-like kinase 3 (Ulk3) in the Sonic hedgehog signaling pathway. J. Biol. Chem. 2010, 285, 30079–30090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloverjan, A.; Piirsoo, M.; Michelson, P.; Kogerman, P.; Osterlund, T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp. Cell Res. 2010, 316, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, H.R.; Martin, M.P.; Luo, Y.; Pireddu, R.; Yang, H.; Gevariya, H.; Ozcan, S.; Zhu, J.Y.; Kendig, R.; Rodriguez, M.; et al. Development of o-chlorophenyl substituted pyrimidines as exceptionally potent aurora kinase inhibitors. J. Med. Chem. 2012, 55, 7392–7416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; DerMardirossian, C.; Bokoch, G.M. Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 2006, 18, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Gohla, A.; Birkenfeld, J.; Bokoch, G.M. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 2005, 7, 21–29. [Google Scholar] [CrossRef]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Wiesner, B.; Baillie, G.S.; Mollajew, R.; Henn, V.; Lorenz, D.; Furkert, J.; Santamaria, K.; Nedvetsky, P.; Hundsrucker, C.; et al. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 2007, 18, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.; Gesbert, F.; Delespine-Carmagnat, M.; Stancou, R.; Pouchelet, M.; Bertoglio, J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996, 15, 510–519. [Google Scholar] [CrossRef]
- Dong, J.M.; Leung, T.; Manser, E.; Lim, L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J. Biol. Chem. 1998, 273, 22554–22562. [Google Scholar] [CrossRef] [Green Version]
- Moon, W.; Matsuzaki, F. Aurora A kinase negatively regulates Rho-kinase by phosphorylation in vivo. Biochem. Biophys. Res. Commun. 2013, 435, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Hannon, G.J. Suppression of p160ROCK bypasses cell cycle arrest after Aurora-A/STK15 depletion. Proc. Natl. Acad. Sci. USA 2004, 101, 8975–8980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; Xiang, J.; Yan, M.; Zhang, Y.; Zhao, Y.; Yue, C.F.; Xu, J.; Zheng, F.M.; Chen, J.N.; Kang, Z.; et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010, 70, 9118–9128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchey, L.; Ottman, R.; Roumanos, M.; Chakrabarti, R. A functional cooperativity between Aurora A kinase and LIM kinase1: Implication in the mitotic process. Cell Cycle 2012, 11, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Maimaiti, Y.; Jie, T.; Jing, Z.; Changwen, W.; Pan, Y.; Chen, C.; Tao, H. Aurora kinase A induces papillary thyroid cancer lymph node metastasis by promoting cofilin-1 activity. Biochem. Biophys. Res. Commun. 2016, 473, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Walter, A.O.; Seghezzi, W.; Korver, W.; Sheung, J.; Lees, E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 2000, 19, 4906–4916. [Google Scholar] [CrossRef] [Green Version]
- de Groot, C.O.; Hsia, J.E.; Anzola, J.V.; Motamedi, A.; Yoon, M.; Wong, Y.L.; Jenkins, D.; Lee, H.J.; Martinez, M.B.; Davis, R.L.; et al. A Cell Biologist’s Field Guide to Aurora Kinase Inhibitors. Front. Oncol. 2015, 5, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Zhu, J.Y.; Lawrence, H.R.; Pireddu, R.; Luo, Y.; Alam, R.; Ozcan, S.; Sebti, S.M.; Lawrence, N.J.; Schönbrunn, E. A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem. Biol. 2012, 7, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903. [Google Scholar] [CrossRef] [Green Version]
- Dodson, C.A.; Kosmopoulou, M.; Richards, M.W.; Atrash, B.; Bavetsias, V.; Blagg, J.; Bayliss, R. Crystal structure of an Aurora-A mutant that mimics Aurora-B bound to MLN8054: Insights into selectivity and drug design. Biochem. J. 2010, 427, 19–28. [Google Scholar] [CrossRef]
- Zorba, A.; Buosi, V.; Kutter, S.; Kern, N.; Pontiggia, F.; Cho, Y.J.; Kern, D. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife 2014, 3, e02667. [Google Scholar] [CrossRef]
- Datta, A.; Yang, C.R.; Limbutara, K.; Chou, C.L.; Rinschen, M.M.; Raghuram, V.; Knepper, M.A. PKA-independent vasopressin signaling in renal collecting duct. FASEB J. 2020, 34, 6129–6146. [Google Scholar] [CrossRef]
- Al-Bataineh, M.M.; Alzamora, R.; Ohmi, K.; Ho, P.Y.; Marciszyn, A.L.; Gong, F.; Li, H.; Hallows, K.R.; Pastor-Soler, N.M. Aurora kinase A activates the vacuolar H+-ATPase (V-ATPase) in kidney carcinoma cells. Am. J. Physiol. Renal. Physiol. 2016, 310, F1216–F1228. [Google Scholar] [CrossRef] [Green Version]
- Bogum, J.; Faust, D.; Zuhlke, K.; Eichhorst, J.; Moutty, M.C.; Furkert, J.; Eldahshan, A.; Neuenschwander, M.; von Kries, J.P.; Wiesner, B.; et al. Small-molecule screening identifies modulators of aquaporin-2 trafficking. J. Am. Soc. Nephrol. 2013, 24, 744–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolin, G.; Tramier, M. Insights into the non-mitotic functions of Aurora kinase A: More than just cell division. Cell. Mol. Life Sci. 2020, 77, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Mori, D.; Yamada, M.; Mimori-Kiyosue, Y.; Shirai, Y.; Suzuki, A.; Ohno, S.; Saya, H.; Wynshaw-Boris, A.; Hirotsune, S. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat. Cell Biol. 2009, 11, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Yool, A.J.; Bill, R.M. Recent breakthroughs and future directions in drugging aquaporins. Trends Pharmacol. Sci. 2022, 43, 30–42. [Google Scholar] [CrossRef]
- Faust, D.; Geelhaar, A.; Eisermann, B.; Eichhorst, J.; Wiesner, B.; Rosenthal, W.; Klussmann, E.; Klussman, E. Culturing primary rat inner medullary collecting duct cells. J. Vis. Exp. 2013, 76, 50366. [Google Scholar] [CrossRef] [Green Version]
- Maric, K.; Oksche, A.; Rosenthal, W. Aquaporin-2 expression in primary cultured rat inner medullary collecting duct cells. Am. J. Physiol. 1998, 275, F796–F801. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Aglio, V.; Tamma, G.; D’Apolito, M.; Addabbo, F.; Procino, G.; Simonetti, M.C.; Montini, G.; Gesualdo, L.; Debler, E.W.; et al. Characterization of Two Novel Missense Mutations in the AQP2 Gene Causing Nephrogenic Diabetes Insipidus. Nephron Physiol. 2007, 105, 33–41. [Google Scholar] [CrossRef]
- Vossenkamper, A.; Nedvetsky, P.I.; Wiesner, B.; Furkert, J.; Rosenthal, W.; Klussmann, E. Microtubules are needed for the perinuclear positioning of aquaporin-2 after its endocytic retrieval in renal principal cells. Am. J. Physiol. Cell Physiol. 2007, 293, C1129–C1138. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Nedvetsky, P.I.; Tabor, V.; Tamma, G.; Beulshausen, S.; Skroblin, P.; Kirschner, A.; Mutig, K.; Boltzen, M.; Petrucci, O.; Vossenkamper, A.; et al. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J. Am. Soc. Nephrol. 2010, 21, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Tamma, G.; Klussmann, E.; Procino, G.; Svelto, M.; Rosenthal, W.; Valenti, G. cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI. J. Cell Sci. 2003, 116, 1519–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Kurz, C.G.; Berger, B.T.; Celik, I.E.; Tjaden, A.; Greco, F.A.; Knapp, S.; Hanke, T. Optimization of pyrazolo[1,5-a]pyrimidines lead to the identification of a highly selective casein kinase 2 inhibitor. Eur. J. Med. Chem. 2020, 208, 112770. [Google Scholar] [CrossRef]
- Robers, M.B.; Dart, M.L.; Woodroofe, C.C.; Zimprich, C.A.; Kirkland, T.A.; Machleidt, T.; Kupcho, K.R.; Levin, S.; Hartnett, J.R.; Zimmerman, K.; et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 2015, 6, 10091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G.; et al. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25, 206–214.e11. [Google Scholar] [CrossRef] [Green Version]
- Christian, F.; Szaszák, M.; Friedl, S.; Drewianka, S.; Lorenz, D.; Goncalves, A.; Furkert, J.; Vargas, C.; Schmieder, P.; Götz, F.; et al. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 2011, 286, 9079–9096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAvoy, T.; Nairn, A.C. Serine/threonine protein phosphatase assays. Curr. Protoc. Mol. Biol. 2010, 92, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Formula | Structure | MW (g/mol) | Target |
|---|---|---|---|---|
| Aurora-A inhibitor I (TC-S 7010) | C31H31ClFN7O2 |  | 588 | AURKA IC50 = 3.4 nM [28] |
| Alisertib (MLN8237) | C27H20ClFN4O4 | 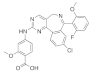 | 519 | AURKA IC50 = 1.2 nM [38] |
| Barasertib-HQPA (AZD1152-HQPA) | C26H30FN7O3 |  | 508 | AURKB IC50 = 0.37 nM [41] |
| Compound Name | Formula | Structure | MW (g/mol) | Target 1 |
|---|---|---|---|---|
| ERJ2-02 [28] | C25H18ClFN5NaO3 | 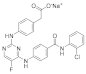 | 513 | AURKA IC50 = 1.32 µM AURKB IC50 > 50 µM |
| ERJ2-03 | C29H26ClFN6O3 | 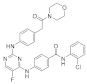 | 560 | AURKA IC50 = 1.08 µM AURKB IC50 > 50 µM |
| ERJ2-04 | C31H31ClFN7O3 | 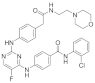 | 603 | AURKA IC50 = 554 nM AURKB IC50 > 50 µM |
| ERJ2-05 | C28H26ClFN6O3 |  | 548 | AURKA IC50 = 239 nM AURKB IC50 = 16.7 µM |
| ERJ2-06 | C25H20ClFN6O2 | 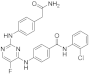 | 490 | AURKA IC50 = 165 nM AURKB IC50 = 13.1 µM |
| ERJ2-07 | C27H24ClFN6O2 | 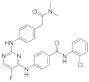 | 518 | Not tested |
| ERJ2-08 | C27H24ClFN6O3 |  | 534 | AURKA IC50 = 237 nM AURKB IC50 = 16.1 µM |
| ERJ2-09 | C18H11FN4Na2O4 | 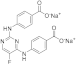 | 412 | AURKA IC50 > 50 µM AURKB IC50 > 50 µM |
| ERJ2-10 | C25H19FN5NaO3 | 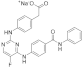 | 479 | AURKA IC50 = 22.8 µM AURKB IC50 > 50 µM |
| ERJ2-11 | C31H32FN7O2 | 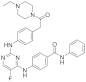 | 553 | AURKA IC50 = 767 nM AURKB IC50 = 42.9 µM |
| ERJ2-12 | C25H26FN6NaO3 |  | 500 | AURKA IC50 > 50 µM AURKB IC50 > 50 µM |
| ERJ2-13 | C31H39FN8O2 | 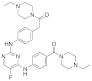 | 574 | AURKA IC50 = 37.3 µM AURKB IC50 > 50 µM |
| ERJ2-15 | C31H38FN7O2 |  | 559 | Not tested |
| ERJ2-16 | C31H39FN8O2 | 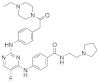 | 574 | AURKA IC50 > 50 µM AURKB IC50 > 50 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baltzer, S.; Bulatov, T.; Schmied, C.; Krämer, A.; Berger, B.-T.; Oder, A.; Walker-Gray, R.; Kuschke, C.; Zühlke, K.; Eichhorst, J.; et al. Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells. Int. J. Mol. Sci. 2022, 23, 763. https://doi.org/10.3390/ijms23020763
Baltzer S, Bulatov T, Schmied C, Krämer A, Berger B-T, Oder A, Walker-Gray R, Kuschke C, Zühlke K, Eichhorst J, et al. Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells. International Journal of Molecular Sciences. 2022; 23(2):763. https://doi.org/10.3390/ijms23020763
Chicago/Turabian StyleBaltzer, Sandrine, Timur Bulatov, Christopher Schmied, Andreas Krämer, Benedict-Tilman Berger, Andreas Oder, Ryan Walker-Gray, Christin Kuschke, Kerstin Zühlke, Jenny Eichhorst, and et al. 2022. "Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells" International Journal of Molecular Sciences 23, no. 2: 763. https://doi.org/10.3390/ijms23020763