
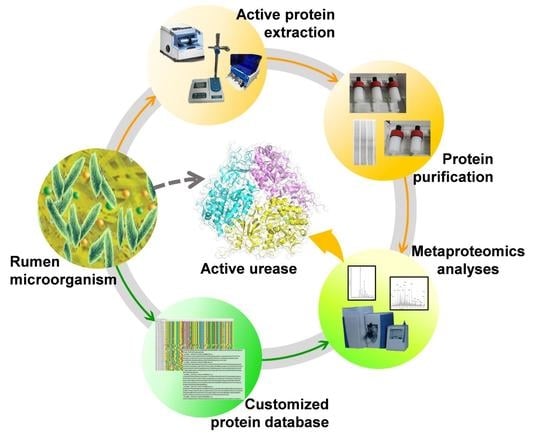
Activity- and Enrichment-Based Metaproteomics Insights into Active Urease from the Rumen Microbiota of Cattle


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Evaluating Extraction Protocols of Active Proteins from Rumen Microbiota
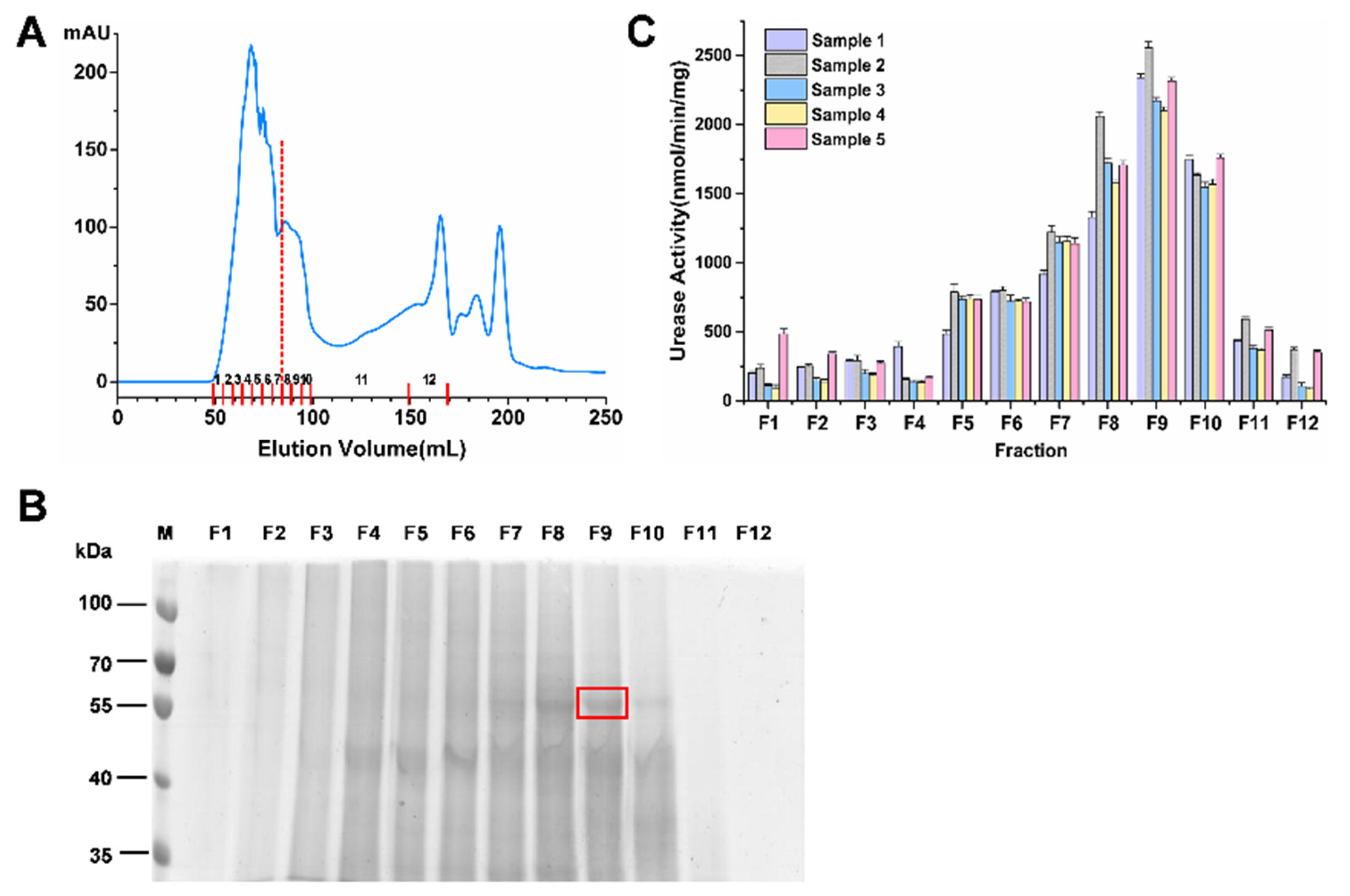
2.2. Enrichment of the Highly Active Urease
2.3. Identification of Rumen Microbial Active Urease
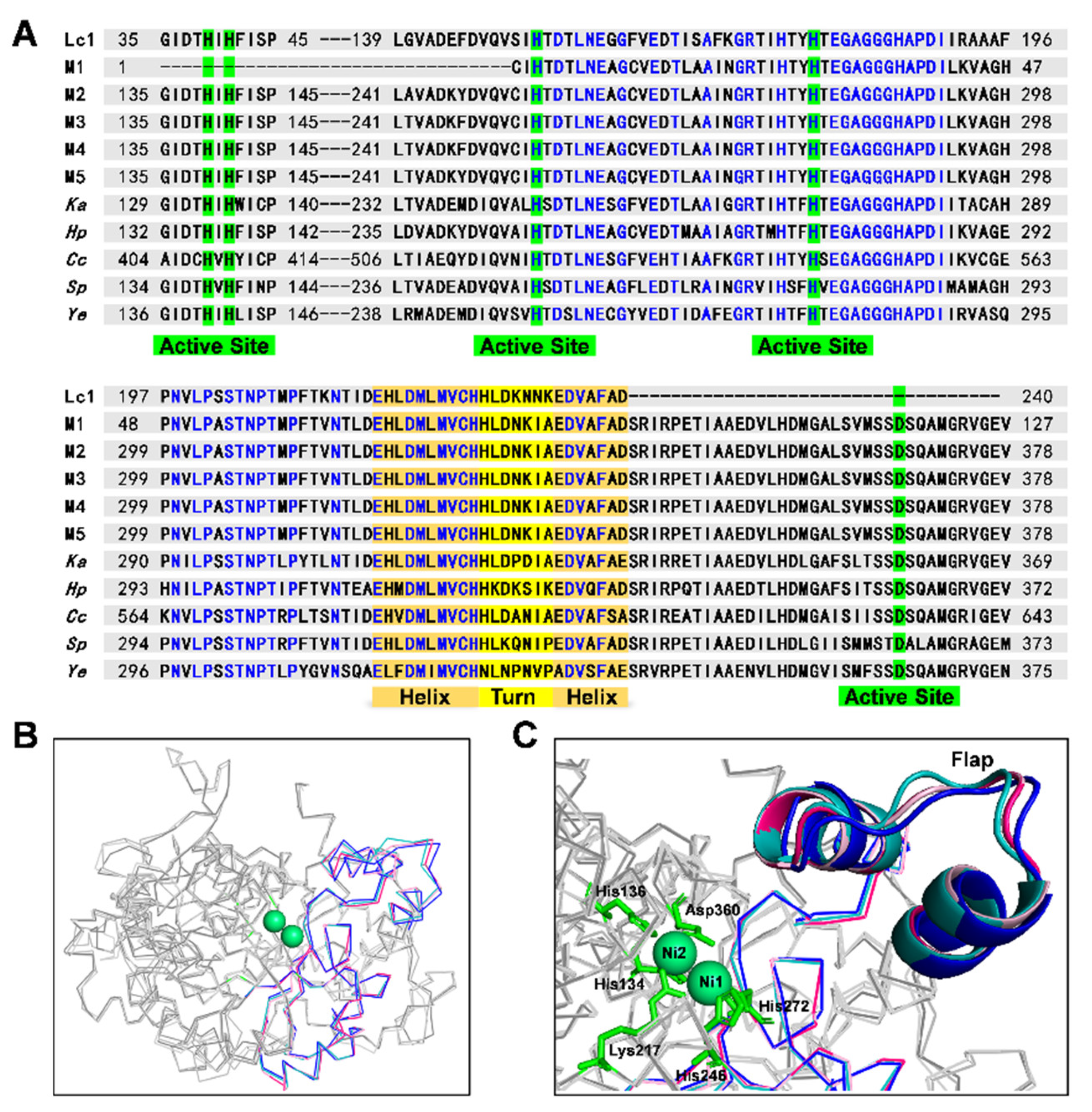
2.4. Comparative Analysis of Identified Rumen Microbial Ureases and the Known Ureases
3. Discussion
4. Materials and Methods
4.1. Collection of Ruminal Microbial Samples
4.2. Extraction of Active Proteins from Rumen Microbiota
4.3. Enrichment of Active Urease by Gel Filtration Chromatography
4.4. LC-MS/MS Analysis
4.5. Metaproteomic Database Search and Urease Identification
4.6. Construction of Phylogenetic Trees for Identified Rumen Microbial Ureases
4.7. Homology Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reynolds, C.K.; Kristensen, N.B. Nitrogen recycling through the gut and the nitrogen economy of ruminants: An asynchronous symbiosis. J. Anim. Sci. 2008, 86, E293–E305. [Google Scholar] [CrossRef] [Green Version]
- Kertz, A.F. Review: Urea feeding to dairy cattle: A historical perspective and review. Prof. Anim. Sci. 2010, 26, 257–272. [Google Scholar] [CrossRef]
- Lin, W.; Mathys, V.; Ang, E.L.; Koh, V.H.; Martínez Gómez, J.M.; Ang, M.L.; Zainul Rahim, S.Z.; Tan, M.P.; Pethe, K.; Alonso, S. Urease activity represents an alternative pathway for Mycobacterium tuberculosis nitrogen metabolism. Infect. Immun. 2012, 80, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Firkins, J.L.; Yu, Z.; Morrison, M. Ruminal nitrogen metabolism: Perspectives for integration of microbiology and nutrition for dairy. J. Dairy Sci. 2007, 1, E1–E16. [Google Scholar] [CrossRef]
- Punj, M.L.; Kakkar, V.K.; Saini, K.S. Studies on the preparation and in vitro breakdown of urea-hemicellulose complex. J. Agric. Sci. 1980, 94, 727–729. [Google Scholar] [CrossRef]
- Gnetegha Ayemele, A.; Ma, L.; Park, T.; Xu, J.; Yu, Z.; Bu, D. Giant milkweed (Calotropis gigantea): A new plant resource to inhibit protozoa and decrease ammoniagenesis of rumen microbiota in vitro without impairing fermentation. Sci. Total Environ. 2020, 743, 140665. [Google Scholar] [CrossRef]
- Abbasi, I.H.R.; Abbasi, F.; Abd El-Hack, M.E.; Abdel-Latif, M.A.; Soomro, R.N.; Hayat, K.; Mohamed, M.A.E.; Bodinga, B.M.; Yao, J.; Cao, Y. Critical analysis of excessive utilization of crude protein in ruminants ration: Impact on environmental ecosystem and opportunities of supplementation of limiting amino acids-a review. Environ. Sci. Pollut. Res. Int. 2018, 25, 181–190. [Google Scholar] [CrossRef]
- Ogawa, J.; Shimizu, S. Microbial enzymes: New industrial applications from traditional screening methods. Trends Biotechnol. 1999, 17, 13–21. [Google Scholar] [CrossRef]
- Patra, A.K.; Aschenbach, J.R. Ureases in the gastrointestinal tracts of ruminant and monogastric animals and their implication in urea-N/ammonia metabolism: A review. J. Adv. Res. 2018, 13, 39–50. [Google Scholar] [CrossRef]
- Michael, S.; Rappé Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar]
- Jin, D.; Zhao, S.; Wang, P.; Zheng, N.; Bu, D.; Beckers, Y.; Wang, J. Insights into Abundant rumen ureolytic bacterial community using rumen simulation system. Front. Microbiol. 2016, 7, 1006. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Zhao, S.; Zheng, N.; Bu, D.; Beckers, Y.; Denman, S.E.; McSweeney, C.S.; Wang, J. Differences in ureolytic bacterial composition between the rumen digesta and rumen wall based on ureC gene classification. Front. Microbiol. 2017, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Hart, E.H.; Creevey, C.J.; Hitch, T.; Kingston-Smith, A.H. Metaproteomics of rumen microbiota indicates niche compartmentalisation and functional dominance in a limited number of metabolic pathways between abundant bacteria. Sci. Rep. 2018, 8, 10504. [Google Scholar] [CrossRef] [Green Version]
- Wooley, J.C.; Godzik, A.; Friedberg, I. A primer on metagenomics. PLoS Comput. Biol. 2010, 6, e1000667. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, P.; Bond, P.L. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004, 6, 911–920. [Google Scholar] [CrossRef]
- Sukul, P.; Schäkermann, S.; Bandow, J.E.; Kusnezowa, A.; Nowrousian, M.; Leichert, L.I. Simple discovery of bacterial biocatalysts from environmental samples through functional metaproteomics. Microbiome 2017, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Speda, J.; Jonsson, B.H.; Carlsson, U.; Karlsson, M. Metaproteomics-guided selection of targeted enzymes for bioprospecting of mixed microbial communities. Biotechnol. Biofuels 2017, 10, 128. [Google Scholar] [CrossRef]
- Beller, H.R.; Rodrigues, A.V.; Zargar, K.; Wu, Y.W.; Saini, A.K.; Saville, R.M.; Pereira, J.H.; Adams, P.D.; Tringe, S.; Petzold, C.J.; et al. Discovery of enzymes for toluene synthesis from anoxic microbial communities. Nat. Chem. Biol. 2018, 14, 451–457. [Google Scholar] [CrossRef]
- Eddhif, B.; Lange, J.; Guignard, N.; Batonneau, Y.; Clarhaut, J.; Papot, S.; Geffroy-Rodier, C.; Poinot, P. Study of a novel agent for TCA precipitated proteins washing—comprehensive insights into the role of ethanol/HCl on molten globule state by multi-spectroscopic analyses. J. Proteom. 2018, 173, 77–88. [Google Scholar] [CrossRef]
- Zargar, K.; Saville, R.; Phelan, R.M.; Tringe, S.G.; Petzold, C.J.; Keasling, J.D.; Beller, H.R. In vitro characterization of phenylacetate decarboxylase, a novel enzyme catalyzing toluene biosynthesis in an anaerobic microbial community. Sci. Rep. 2016, 6, 31362. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. Mass spectrometrists should search only for peptides they care about. Nat. Methods 2015, 12, 605–608. [Google Scholar] [CrossRef] [Green Version]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Barnouin, K. Guidelines for experimental design and data analysis of proteomic mass spectrometry-based experiments. Amino Acids 2011, 40, 259–260. [Google Scholar] [CrossRef]
- Muth, T.; Kolmeder, C.A.; Salojarvi, J.; Keskitalo, S.; Varjosalo, M.; Verdam, F.J.; Rensen, S.S.; Reichl, U.; de Vos, W.M.; Rapp, E.; et al. Navigating through metaproteomics data: A logbook of database searching. Proteomics 2015, 15, 3439–3453. [Google Scholar] [CrossRef]
- Jagtap, P.; Goslinga, J.; Kooren, J.A.; McGowan, T.; Wroblewski, M.S.; Seymour, S.L.; Griffin, T.J. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 2013, 13, 1352–1357. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Tanca, A.; Jia, B.; Yang, R.; Wang, B.; Zhang, Y.; Li, J. Metagenomic taxonomy-guided database-searching strategy for improving metaproteomic analysis. J. Proteome Res. 2018, 17, 1596–1605. [Google Scholar] [CrossRef]
- Tang, H.; Li, S.; Ye, Y. A graph-centric approach for metagenome-guided peptide and protein identification in metaproteomics. PLoS Comput. Biol. 2016, 12, e1005224. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Gonnelli, G.; Stock, M.; Verwaeren, J.; Maddelein, D.; De Baets, B.; Martens, L.; Degroeve, S. A decoy-free approach to the identification of peptides. J. Proteome Res. 2015, 14, 1792–1798. [Google Scholar] [CrossRef]
- Wozny, M.A.; Bryant, M.P.; Holdeman, L.V.; Moore, W.E.C. Urease assay and ureaseproducing species of anaerobes in the bovine rumen and human feces. Appl. Microbiol. 1977, 33, 1097–1104. [Google Scholar]
- Huws, S.A.; Edwards, J.E.; Creevey, C.J.; Rees Stevens, P.; Lin, W.; Girdwood, S.E.; Pachebat, J.A.; Kingston-Smith, A.H. Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass. FEMS Microbiol. Ecol. 2016, 92, fiv137. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.P.; McCormick, C.A.; Suen, G. Fibrobacter communities in the gastrointestinal tracts of diverse hindgut-fermenting herbivores are distinct from those of the rumen. Environ. Microbiol. 2017, 19, 3768–3783. [Google Scholar] [CrossRef]
- Weatherburn, M. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 1967, 39, 971–973. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, S.; He, Y.; Zheng, N.; Yan, X.; Wang, J. Pipeline for targeted metaproteomic analyses to assess the diversity of cattle rumen microbial urease. Front. Microbiol. 2020, 11, 573414. [Google Scholar] [CrossRef]
- Minas, K.; McEwan, N.R.; Newbold, C.J.; Scott, K.P. Optimizationofahigh-throughput CTAB-based protocol for the extraction of qPCR-grade DNA from rumen fluid, plant and bacterial pure cultures. FEMS Microbiol. Lett. 2011, 325, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Xiong, Z.; Li, M.; Zheng, N.; Zhao, S.; Wang, J. Activity- and Enrichment-Based Metaproteomics Insights into Active Urease from the Rumen Microbiota of Cattle. Int. J. Mol. Sci. 2022, 23, 817. https://doi.org/10.3390/ijms23020817
Zhang X, Xiong Z, Li M, Zheng N, Zhao S, Wang J. Activity- and Enrichment-Based Metaproteomics Insights into Active Urease from the Rumen Microbiota of Cattle. International Journal of Molecular Sciences. 2022; 23(2):817. https://doi.org/10.3390/ijms23020817
Chicago/Turabian StyleZhang, Xiaoyin, Zhanbo Xiong, Ming Li, Nan Zheng, Shengguo Zhao, and Jiaqi Wang. 2022. "Activity- and Enrichment-Based Metaproteomics Insights into Active Urease from the Rumen Microbiota of Cattle" International Journal of Molecular Sciences 23, no. 2: 817. https://doi.org/10.3390/ijms23020817