
HDAC2 Is Involved in the Regulation of BRN3A in Melanocytes and Melanoma

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. BRN3A Expression in Melanocytes Is Not Silenced by DNA Hypermethylation
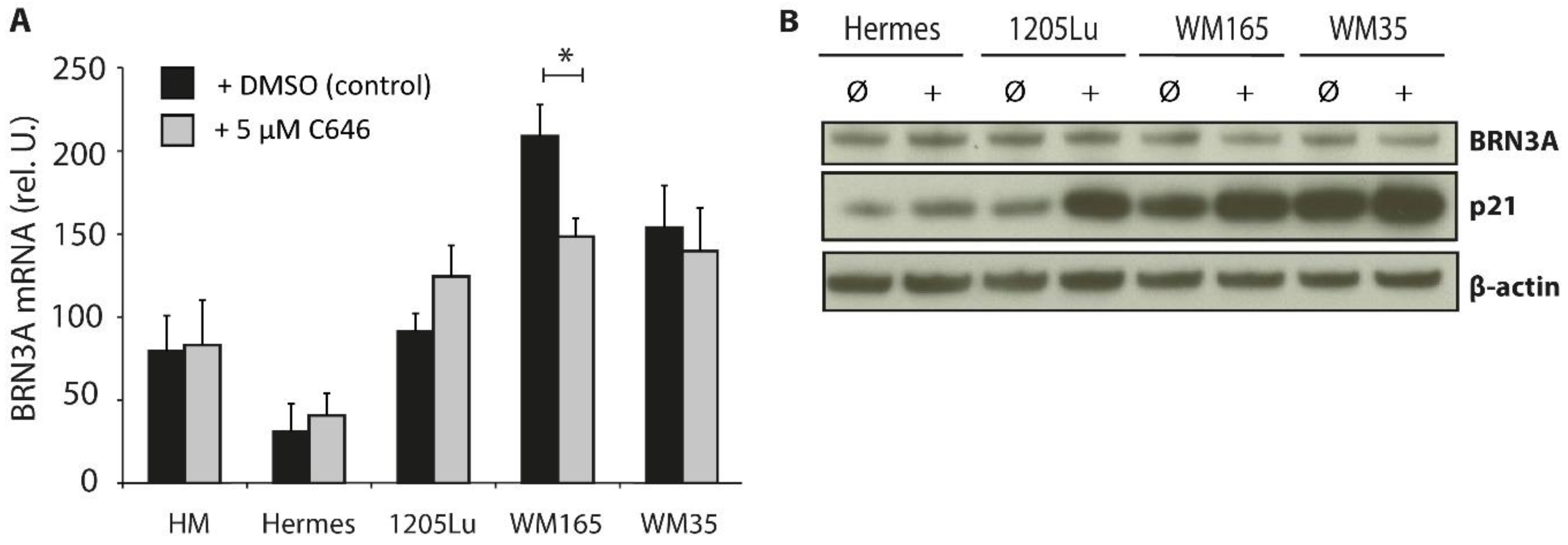
2.2. Influence of HAT Inhibition on BRN3A Expression Levels
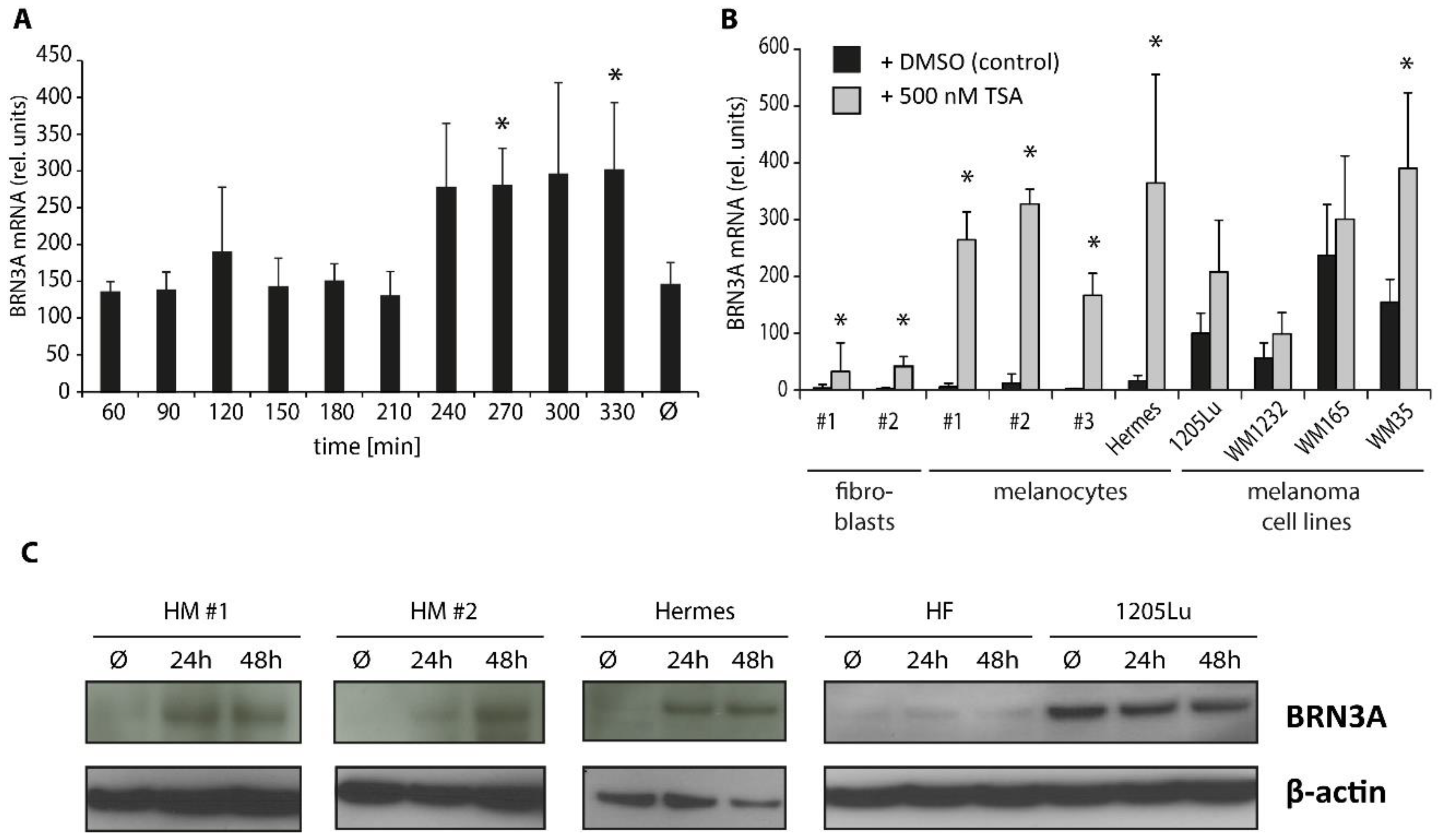
2.3. Trichostatin A Induces BRN3A Expression in Melanocytes and Melanoma Cell Lines
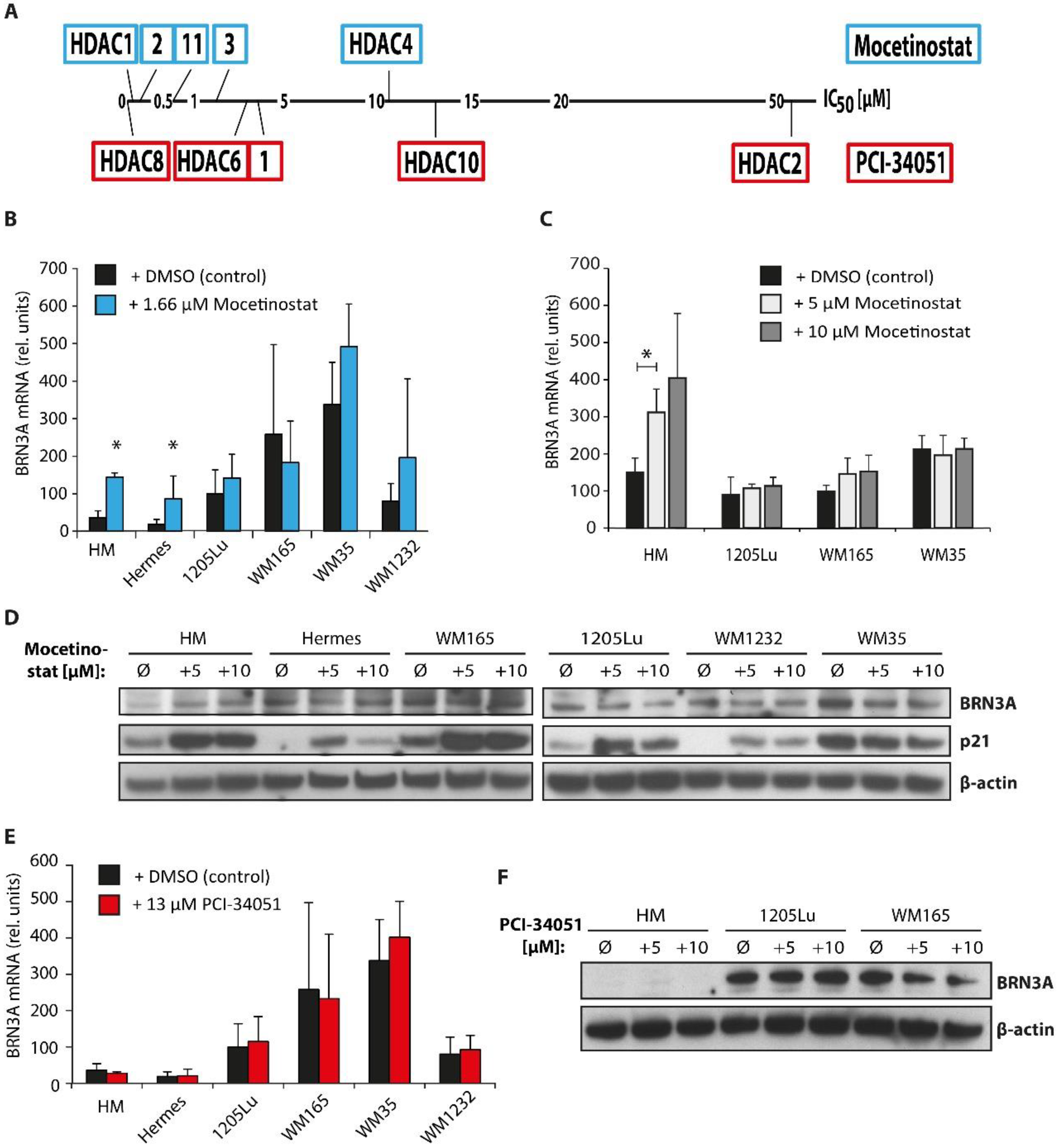
2.4. Class I HDAC Inhibition Increases BRN3A Expression in Melanocytes
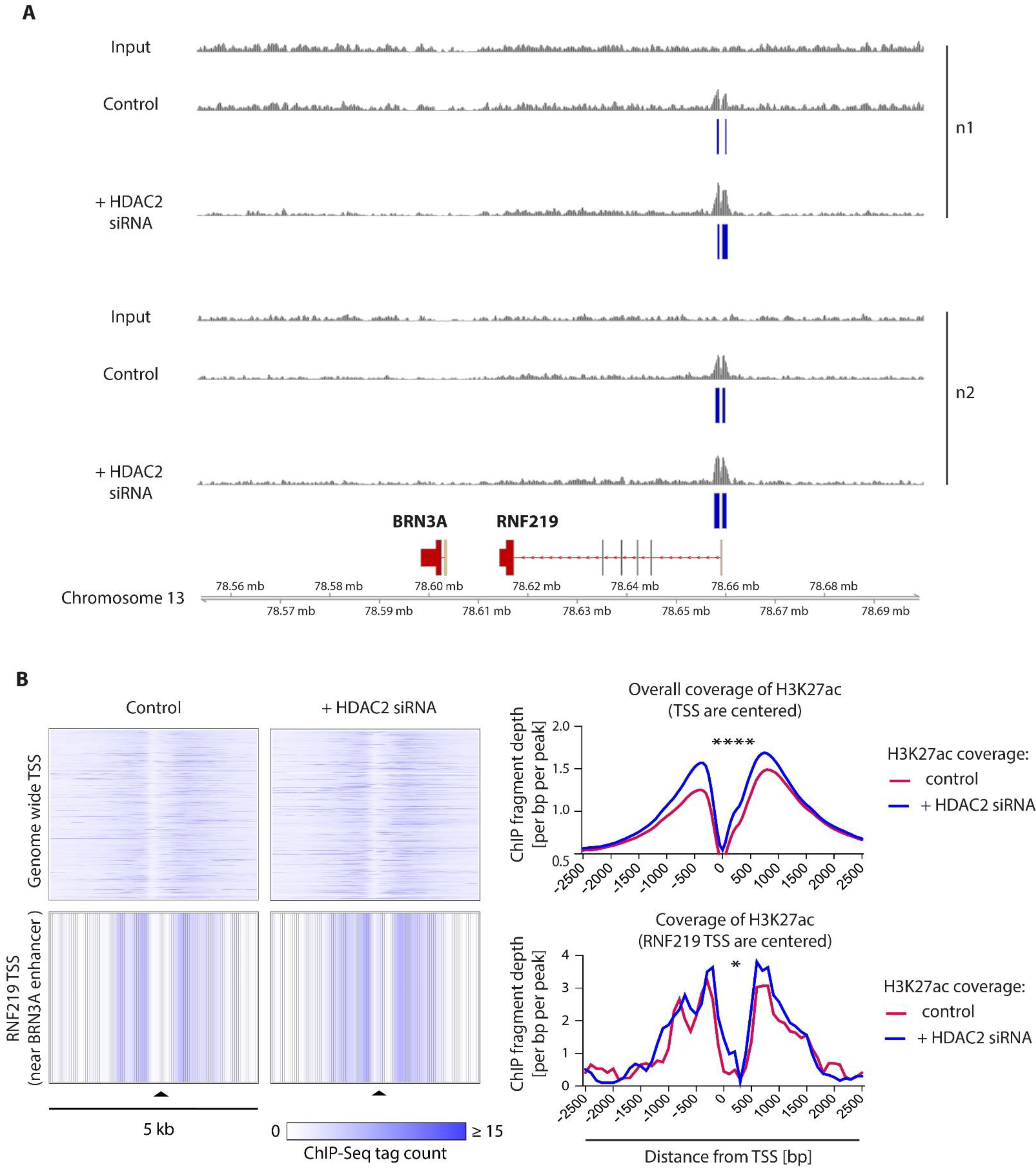
2.5. HDAC2 Is a Key Epigenetic Regulator of BRN3A in Melanocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Pharmacologic DNMT, HAT, and HDAC Inhibition
4.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.4. Protein Extraction and Immunoblotting
4.5. DNA Methylation Status Analysis: Bisulfite Conversion, Methylation PCR, and Sequencing
4.6. siRNA Transfection
4.7. Ectopic HDAC2 Expression
4.8. Chromatin Immunoprecipitation and Sequencing (ChIP-Seq), Mapping, and Data Analysis
4.9. Light Microscopy
4.10. Cell Viability Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Crane, J.F.; Trainor, P.A. Neural crest stem and progenitor cells. Annu. Rev. Cell Dev. Biol. 2006, 22, 267–286. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tyminska, A. Skin melanocytes: Biology and development. Postepy Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besch, R.; Berking, C. POU transcription factors in melanocytes and melanoma. Eur. J. Cell Biol. 2014, 93, 55–60. [Google Scholar] [CrossRef]
- Fedtsova, N.G.; Turner, E.E. Brn-3.0 expression identifies early post-mitotic CNS neurons and sensory neural precursors. Mech. Dev. 1995, 53, 291–304. [Google Scholar] [CrossRef]
- McEvilly, R.J.; Erkman, L.; Luo, L.; Sawchenko, P.E.; Ryan, A.F.; Rosenfeld, M.G. Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature 1996, 384, 574–577. [Google Scholar] [CrossRef]
- Xiang, M.; Gan, L.; Zhou, L.; Klein, W.H.; Nathans, J. Targeted deletion of the mouse POU domain gene Brn-3a causes selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proc. Natl. Acad. Sci. USA 1996, 93, 11950–11955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budhram-Mahadeo, V.; Ndisang, D.; Ward, T.; Weber, B.L.; Latchman, D.S. The Brn-3b POU family transcription factor represses expression of the BRCA-1 anti-oncogene in breast cancer cells. Oncogene 1999, 18, 6684–6691. [Google Scholar] [CrossRef] [Green Version]
- Collum, R.G.; Fisher, P.E.; Datta, M.; Mellis, S.; Thiele, C.; Huebner, K.; Croce, C.M.; Israel, M.A.; Theil, T.; Moroy, T.; et al. A novel POU homeodomain gene specifically expressed in cells of the developing mammalian nervous system. Nucleic Acids Res. 1992, 20, 4919–4925. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Treacy, M.N.; Simmons, D.M.; Ingraham, H.A.; Swanson, L.W.; Rosenfeld, M.G. Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature 1989, 340, 35–41. [Google Scholar] [CrossRef]
- Hohenauer, T.; Berking, C.; Schmidt, A.; Haferkamp, S.; Senft, D.; Kammerbauer, C.; Fraschka, S.; Graf, S.A.; Irmler, M.; Beckers, J.; et al. The neural crest transcription factor Brn3a is expressed in melanoma and required for cell cycle progression and survival. EMBO Mol. Med. 2013, 5, 919–934. [Google Scholar] [CrossRef]
- Diss, J.K.; Faulkes, D.J.; Walker, M.M.; Patel, A.; Foster, C.S.; Budhram-Mahadeo, V.; Djamgoz, M.B.; Latchman, D.S. Brn-3a neuronal transcription factor functional expression in human prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 83–91. [Google Scholar] [CrossRef]
- Ndisang, D.; Budhram-Mahadeo, V.; Latchman, D.S. The Brn-3a transcription factor plays a critical role in regulating human papilloma virus gene expression and determining the growth characteristics of cervical cancer cells. J. Biol. Chem. 1999, 274, 28521–28527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Latifi, A.; Riley, C.B.; Findlay, J.K.; Quinn, M.A. Neuronal transcription factor Brn-3a(l) is over expressed in high-grade ovarian carcinomas and tumor cells from ascites of patients with advanced-stage ovarian cancer. J. Ovarian Res. 2010, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell-Price, J.; Clark, A.J.; King, P. DNA methylation and silencing of gene expression. Trends Endocrinol. Metab. 2000, 11, 142–148. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Hornig, E.; Heppt, M.V.; Graf, S.A.; Ruzicka, T.; Berking, C. Inhibition of histone deacetylases in melanoma-a perspective from bench to bedside. Exp. Dermatol. 2016, 25, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Dahiya, R. MethPrimer CpG Island Prediction Tool. Available online: http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi (accessed on 22 January 2019).
- Issa, J.P. Decitabine. Curr. Opin. Oncol. 2003, 15, 446–451. [Google Scholar] [CrossRef]
- Zhu, W.G.; Hileman, T.; Ke, Y.; Wang, P.; Lu, S.; Duan, W.; Dai, Z.; Tong, T.; Villalona-Calero, M.A.; Plass, C.; et al. 5-aza-2’-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J. Biol. Chem. 2004, 279, 15161–15166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar, O.; Achberger, S.; Aldrich, W.; Hu, Z.; Negrotto, S.; Saunthararajah, Y.; Triozzi, P. Epigenetic regulation by decitabine of melanoma differentiation in vitro and in vivo. Int. J. Cancer 2012, 131, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Howell, P.; Ren, S.; Fodstad, O.; Riker, A.I. The 14-3-3sigma gene promoter is methylated in both human melanocytes and melanoma. BMC Cancer 2009, 9, 162. [Google Scholar] [CrossRef] [Green Version]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Eller, M.S.; Elm, C.; Larocca, C.A.; Ryu, B.; Panova, I.P.; Dancy, B.M.; Bowers, E.M.; Meyers, D.; Lareau, L.; et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J. Investig. Dermatol. 2013, 133, 2444–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Horinouchi, S.; Beppu, T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 1995, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Kiviharju, T.M.; Jarvinen, P.M.; Ra, R.; Laiho, M. Melanoma cell lines are susceptible to histone deacetylase inhibitor TSA provoked cell cycle arrest and apoptosis. Pigment Cell Res. 2005, 18, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.T.; Trachy-Bourget, M.C.; Kalita, A.; Liu, J.; Lu, A.H.; Zhou, N.Z.; Robert, M.F.; et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Tsai, S.C.; Seto, E. Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GeneCards. POU Class 4 Homeobox 1 (Brn3a) GeneCard. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=pou4f1 (accessed on 18 September 2019).
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsman, D.G.; Kobel, M. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Marquard, L.; Gjerdrum, L.M.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Woan, K.; Cheng, F.; Wang, H.; Perez-Villarroel, P.; Lee, C.; Lienlaf, M.; Atadja, P.; Seto, E.; Weber, J.; et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013, 23, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.; Fan, K.; Paulson, K.G.; Nghiem, P.; Schrama, D.; Becker, J.C. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 2016, 6, 21678. [Google Scholar] [CrossRef] [Green Version]
- Lai, F.; Jin, L.; Gallagher, S.; Mijatov, B.; Zhang, X.D.; Hersey, P. Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv. Pharmacol. 2012, 65, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef]
- Coulombe, P.; Nassar, J.; Peiffer, I.; Stanojcic, S.; Sterkers, Y.; Delamarre, A.; Bocquet, S.; Mechali, M. The ORC ubiquitin ligase OBI1 promotes DNA replication origin firing. Nat. Commun. 2019, 10, 2426. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Farydak (Panobinostat), Summary of the European Public Assessment Report by EMA. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/farydak (accessed on 18 January 2019).
- Berking, C.; Takemoto, R.; Satyamoorthy, K.; Elenitsas, R.; Herlyn, M. Basic fibroblast growth factor and ultraviolet B transform melanocytes in human skin. Am. J. Pathol. 2001, 158, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Sviderskaya, E.V.; Gray-Schopfer, V.C.; Hill, S.P.; Smit, N.P.; Evans-Whipp, T.J.; Bond, J.; Hill, L.; Bataille, V.; Peters, G.; Kipling, D.; et al. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: Possible implications for melanoma progression. J. Natl. Cancer Inst. 2003, 95, 723–732. [Google Scholar] [CrossRef]
- Roche. Universal ProbeLibrary Assay Design Center. Available online: https://lifescience.roche.com/en_de/brands/universal-probe-library.html#assay-design-center (accessed on 18 July 2019).
- Matheis, F.; Heppt, M.V.; Graf, S.A.; Duwell, P.; Kammerbauer, C.; Aigner, A.; Besch, R.; Berking, C. A bifunctional approach of immunostimulation and uPAR inhibition shows potent antitumor activity in melanoma. J. Investig. Dermatol. 2016, 136, 2475–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besch, R.; Poeck, H.; Hohenauer, T.; Senft, D.; Hacker, G.; Berking, C.; Hornung, V.; Endres, S.; Ruzicka, T.; Rothenfusser, S.; et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Investig. 2009, 119, 2399–2411. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information (NCBI). Basic Local Alignment Search Tool (BLAST®). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 25 July 2019).
- Graf, S.A.; Heppt, M.V.; Wessely, A.; Krebs, S.; Kammerbauer, C.; Hornig, E.; Strieder, A.; Blum, H.; Bosserhoff, A.K.; Berking, C. The myelin protein PMP2 is regulated by SOX10 and drives melanoma cell invasion. Pigment Cell Melanoma Res. 2018, 32, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Kappelmann-Fenzl, M.; Gebhard, C.; Matthies, A.O.; Kuphal, S.; Rehli, M.; Bosserhoff, A.K. C-Jun drives melanoma progression in PTEN wild type melanoma cells. Cell Death Dis. 2019, 10, 584. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef] [PubMed]
- Benner, C. HOMER: Software for Motif Discovery and Next Generation Sequencing Analysis. Available online: http://homer.ucsd.edu/homer/ (accessed on 10 September 2019).
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RStudio. RStudio Software. Available online: https://www.rstudio.com/ (accessed on 10 September 2019).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Forward (5′→3′) | Primer Reverse (5′→3′) | Probe No. |
|---|---|---|---|
| HPRT | TGACCTTGATTTATTTTGCATACC | CGAGCAAGACGTTCAGTCCT | 73 |
| BRN3A | TTTCCTCCACCCATTCTCTG | ACCCCAGTCCTCAAGGCTA | 56 |
| HDAC2 | CAGATCGTGTAATGACGGTATCA | CCTTTTCCAGCACCAATATCC | 72 |
| CDKN1A | TCACTGTCTTGTACCCTTGTGC | GGCGTTTGGAGTGGTAGAAA | 32 |
| SFN | TCGTAGGAATTGAGGAGTGTCC | ACACACACACCCATCTCCAG | 5 |
| Cycle Step | Temperature | Time | |
|---|---|---|---|
| Initial denaturation | 95 °C | 60 s | 1 cycle |
| Denaturation | 95 °C | 30 s | |
| Annealing | 53 °C | 30 s | 40 cycles |
| Extension | 68 °C | 30 s | |
| Final extension | 68 °C | 5 min | 1 cycle |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heppt, M.V.; Wessely, A.; Hornig, E.; Kammerbauer, C.; Graf, S.A.; Besch, R.; French, L.E.; Matthies, A.; Kuphal, S.; Kappelmann-Fenzl, M.; et al. HDAC2 Is Involved in the Regulation of BRN3A in Melanocytes and Melanoma. Int. J. Mol. Sci. 2022, 23, 849. https://doi.org/10.3390/ijms23020849
Heppt MV, Wessely A, Hornig E, Kammerbauer C, Graf SA, Besch R, French LE, Matthies A, Kuphal S, Kappelmann-Fenzl M, et al. HDAC2 Is Involved in the Regulation of BRN3A in Melanocytes and Melanoma. International Journal of Molecular Sciences. 2022; 23(2):849. https://doi.org/10.3390/ijms23020849
Chicago/Turabian StyleHeppt, Markus V., Anja Wessely, Eva Hornig, Claudia Kammerbauer, Saskia A. Graf, Robert Besch, Lars E. French, Alexander Matthies, Silke Kuphal, Melanie Kappelmann-Fenzl, and et al. 2022. "HDAC2 Is Involved in the Regulation of BRN3A in Melanocytes and Melanoma" International Journal of Molecular Sciences 23, no. 2: 849. https://doi.org/10.3390/ijms23020849