
Therapeutic CFTR Correction Normalizes Systemic and Lung-Specific S1P Level Alterations Associated with Heart Failure


Abstract
1. Introduction
2. Results
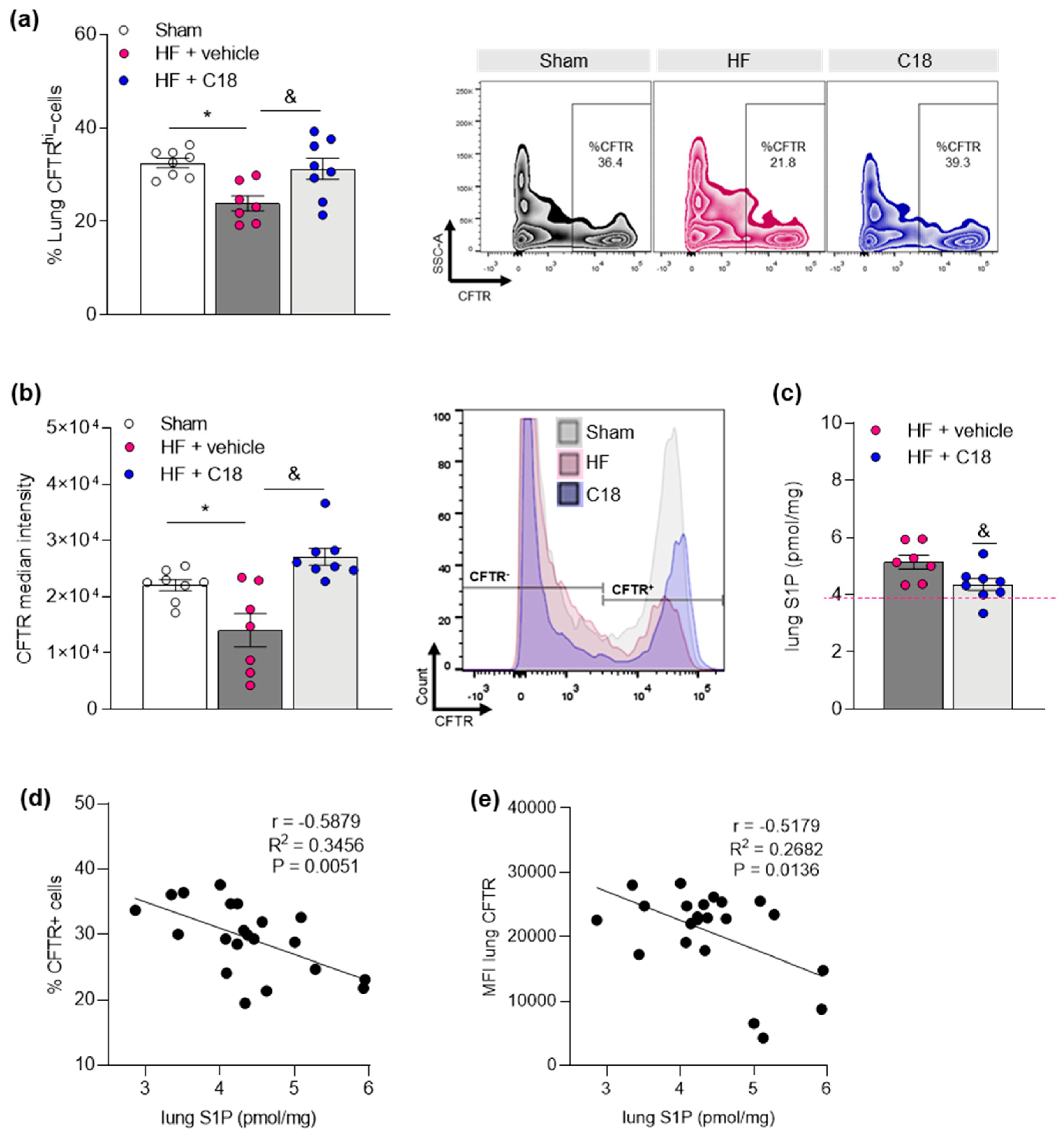
2.1. Impaired Pulmonary CFTR Expression during Heart Failure Links to Increased Sphingosine-1-Phosphate Concentrations in the Lung
2.2. Therapeutic CFTR Correction Attenuates Heart Failure-Associated Sphingosine-1-Phosphate Level Elevation in the Lung
2.3. Therapeutic CFTR Correction Normalizes Heart Failure-Associated Elevation of Plasma Sphingosine-1-Phosphate Levels with Implications for Systemic Inflammation
2.4. Therapeutic CFTR Correction Attenuates Heart Failure-Associated Pulmonary Inflammation
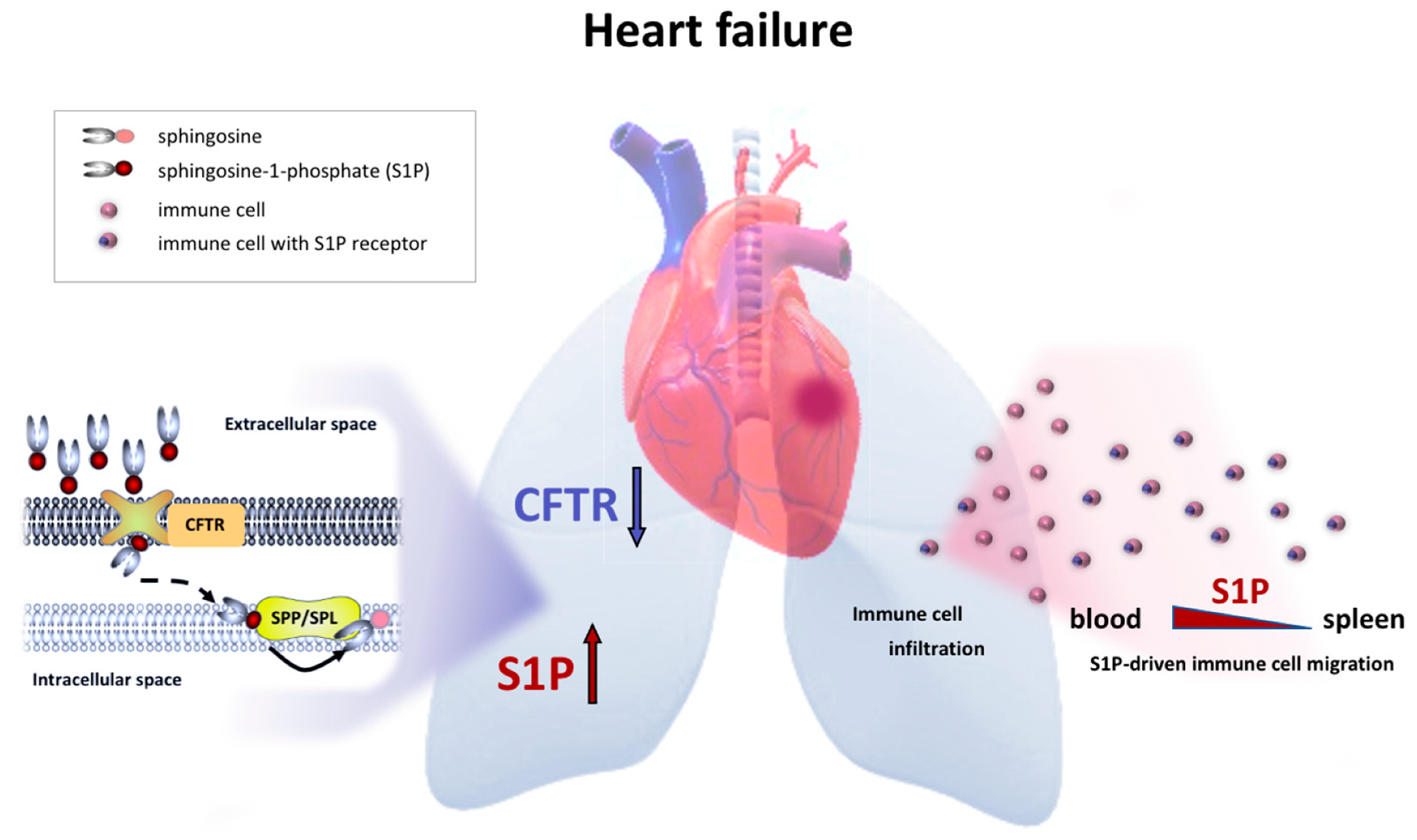
3. Discussion
4. Conclusions
5. Future Perspectives
6. Materials and Methods
6.1. Chemicals and Reagents
6.2. Animals
6.3. Induction of Myocardial Infarction in Mice
6.4. Assessment of Cardiac Function Using Magnetic Resonance Imaging
6.5. Fluorescence Activated Cell Sorting
6.6. Western Blotting
6.7. Quantitative Real-Time PCR (qPCR)
6.8. S1P Mass Spectrometry
6.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Ordernr | Concentration and Application | Size [kDa] |
|---|---|---|---|---|
| Live/Dead Aqua | Invitrogen | L34966 | 1:500 FACS | |
| B220 AF488 | R&D | FAB1217G | 1:200 FACS | - |
| CD11b PE Tex Red | Invitrogen | RM2817 | 1:200 FACS | - |
| CD3 eFluor450 | eBioscience | 48-0032-82 | 1:200 FACS | - |
| CD45 AF700 | R&D | FAB114N | 1:200 FACS | - |
| CFTR | Thermo Fisher | MA1-935 | 1:400 FACS; 1:1000 WB | 170 |
| HRP-labeled goat anti-mouse | Cell signalling | 7076S | 1:10,000 WB | - |
| Ly6C PE-Cy7 | eBioscience | 25-5932-82 | 1:200 FACS | - |
| Ly6G APC | eBioscience | 17-9668-82 | 1:200 FACS | - |
| S1P1 PE | R&D | FAB7089P | 1:200 FACS | - |
| Gene | Sequence (5′ to 3′) | Annealing Temp. [°C] |
|---|---|---|
| L14 | Fw: GGCTTTAGTGGATGGACCCT Rv: ATTGATATCCGCCTTCTCCC | 59.4 57.3 |
| SphK1 | Fw: GGTGAATGGGCTAATGGAACG Rv: CTGCTCGTACCCAGCATAGTG | 59.8 61.8 |
| SphK2 | Fw: CACGGCGAGTTTGGTTCCTA Rv: CTTCTGGCTTTGGGCGTAGT | 59.4 59.4 |
| Sgpl1 | Fw: GGTGTATGAGCTTATCTTCCAGC Rv: CTGTTGTTCGATCTTACGTCCA | 60.6 58.4 |
| Sgpp1 | Fw: GAGCAACTTGCCGCTCTACTA Rv: GGTCGAGATTCCAGATCCAGAA | 59.8 60.3 |
| Sgpp2 | Fw: GTTCTCTACGCTGGTGTGTCT Rv: GCAGGGTAGGTCAGAGCAAT | 59.8 59.4 |
| Il1b | Fw: GAAGAGCCCATCCTCTGTGA Rv: TTCATCTCGGAGCCTGTAGTG | 59.4 59.8 |
References
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Eken, A.; Duhen, R.; Singh, A.K.; Fry, M.; Buckner, J.H.; Kita, M.; Bettelli, E.; Oukka, M. S1P1 deletion differentially affects TH17 and Regulatory T cells. Sci. Rep. 2017, 7, 12905. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, J.; Azam, M.A.; Kroetsch, J.T.; Leong-Poi, H.; Momen, M.A.; Voigtlaender-Bolz, J.; Scherer, E.Q.; Meissner, A.; Bolz, S.S.; Husain, M. Sphingosine-1-phosphate-dependent activation of p38 MAPK maintains elevated peripheral resistance in heart failure through increased myogenic vasoconstriction. Circ. Res. 2010, 107, 923–933. [Google Scholar] [CrossRef]
- Cantalupo, A.; Gargiulo, A.; Dautaj, E.; Liu, C.; Zhang, Y.; Hla, T.; Di Lorenzo, A. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 2017, 70, 426–434. [Google Scholar] [CrossRef]
- Yang, J.; Noyan-Ashraf, M.H.; Meissner, A.; Voigtlaender-Bolz, J.; Kroetsch, J.T.; Foltz, W.; Jaffray, D.; Kapoor, A.; Momen, A.; Heximer, S.P.; et al. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation 2012, 126, 196–206. [Google Scholar] [CrossRef]
- Meissner, A.; Miro, F.; Jimenez-Altayo, F.; Jurado, A.; Vila, E.; Planas, A.M. Sphingosine-1-phosphate signalling-a key player in the pathogenesis of Angiotensin II-induced hypertension. Cardiovasc. Res. 2017, 113, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Yagi, K.; Lidington, D.; Wan, H.; Fares, J.C.; Meissner, A.; Sumiyoshi, M.; Ai, J.; Foltz, W.D.; Nedospasov, S.A.; Offermanns, S.; et al. Therapeutically Targeting Tumor Necrosis Factor-alpha/Sphingosine-1-Phosphate Signaling Corrects Myogenic Reactivity in Subarachnoid Hemorrhage. Stroke 2015, 46, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Lidington, D.; Fares, J.C.; Uhl, F.E.; Dinh, D.D.; Kroetsch, J.T.; Sauvé, M.; Malik, F.A.; Matthes, F.; Vanherle, L.; Adel, A.; et al. CFTR Therapeutics Normalize Cerebral Perfusion Deficits in Mouse Models of Heart Failure and Subarachnoid Hemorrhage. JACC Basic Trans. Sci. 2019, 4, 940–958. [Google Scholar] [CrossRef]
- Obinata, H.; Kuo, A.; Wada, Y.; Swendeman, S.; Liu, C.H.; Blaho, V.A.; Nagumo, R.; Satoh, K.; Izumi, T.; Hla, T. Identification of ApoA4 as a sphingosine 1-phosphate chaperone in ApoM- and albumin-deficient mice. J. Lipid Res. 2019, 60, 1912–1921. [Google Scholar] [CrossRef]
- Salas-Perdomo, A.; Miro-Mur, F.; Gallizioli, M.; Brait, V.H.; Justicia, C.; Meissner, A.; Urra, X.; Chamorro, A.; Planas, A.M. Role of the S1P pathway and inhibition by fingolimod in preventing hemorrhagic transformation after stroke. Sci. Rep. 2019, 9, 8309. [Google Scholar] [CrossRef]
- Don-Doncow, N.; Vanherle, L.; Zhang, Y.; Meissner, A. T-Cell Accumulation in the Hypertensive Brain: A Role for Sphingosine-1-Phosphate-Mediated Chemotaxis. Int. J. Mol. Sci. 2019, 20, 537. [Google Scholar] [CrossRef]
- Siedlinski, M.; Nosalski, R.; Szczepaniak, P.; Ludwig-Galezowska, A.H.; Mikolajczyk, T.; Filip, M.; Osmenda, G.; Wilk, G.; Nowak, M.; Wolkow, P.; et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci. Rep. 2017, 7, 44131. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine Kinase 1: A Potential Therapeutic Target in Pulmonary Arterial Hypertension? Trends Mol. Med. 2017, 23, 786–798. [Google Scholar] [CrossRef]
- Bot, M.; Van Veldhoven, P.P.; de Jager, S.C.; Johnson, J.; Nijstad, N.; Van Santbrink, P.J.; Westra, M.M.; Van Der Hoeven, G.; Gijbels, M.J.; Muller-Tidow, C.; et al. Hematopoietic sphingosine 1-phosphate lyase deficiency decreases atherosclerotic lesion development in LDL-receptor deficient mice. PLoS ONE 2013, 8, e63360. [Google Scholar] [CrossRef]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef]
- Polzin, A.; Piayda, K.; Keul, P.; Dannenberg, L.; Mohring, A.; Graler, M.; Zeus, T.; Kelm, M.; Levkau, B. Plasma sphingosine-1-phosphate concentrations are associated with systolic heart failure in patients with ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 110, 35–37. [Google Scholar] [CrossRef]
- Jujic, A.; Matthes, F.; Vanherle, L.; Petzka, H.; Orho-Melander, M.; Nilsson, P.M.; Magnusson, M.; Meissner, A. Plasma S1P (Sphingosine-1-Phosphate) Links to Hypertension and Biomarkers of Inflammation and Cardiovascular Disease: Findings From a Translational Investigation. Hypertension 2021, 78, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sugimoto, K.; Cao, Y.; Mori, M.; Guo, L.; Tan, G. Serum Sphingosine 1-Phosphate (S1P): A Novel Diagnostic Biomarker in Early Acute Ischemic Stroke. Front. Neurol. 2020, 11, 985. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Duner, P.; Stahlman, M.; Mollet, I.G.; Asciutto, G.; Grufman, H.; Nitulescu, M.; Persson, A.F.; Fisher, R.M.; Melander, O.; et al. Sphingolipids Contribute to Human Atherosclerotic Plaque Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Gowda, S.G.B.; Gowda, D.; Kain, V.; Chiba, H.; Hui, S.P.; Chalfant, C.E.; Parcha, V.; Arora, P.; Halade, G.V. Sphingosine-1-phosphate interactions in the spleen and heart reflect extent of cardiac repair in mice and failing human hearts. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H599–H611. [Google Scholar] [CrossRef]
- Lidington, D.; Peter, B.F.; Meissner, A.; Kroetsch, J.T.; Pitson, S.M.; Pohl, U.; Bolz, S.S. The phosphorylation motif at serine 225 governs the localization and function of sphingosine kinase 1 in resistance arteries. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1916–1922. [Google Scholar] [CrossRef]
- Meissner, A.; Yang, J.; Kroetsch, J.T.; Sauve, M.; Dax, H.; Momen, A.; Noyan-Ashraf, M.H.; Heximer, S.; Husain, M.; Lidington, D.; et al. Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation 2012, 125, 2739–2750. [Google Scholar] [CrossRef]
- Boujaoude, L.C.; Bradshaw-Wilder, C.; Mao, C.; Cohn, J.; Ogretmen, B.; Hannun, Y.A.; Obeid, L.M. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: Modulation of cellular activity of sphingosine 1-phosphate. J. Biol. Chem. 2001, 276, 35258–35264. [Google Scholar] [CrossRef]
- Uhl, F.; Vanherle, L.; Meissner, A. Cystic Fibrosis Transmembrane Regulator Correction Attenuates Heart Failure-Induced Lung Inflammation. Authorea 2021. [Google Scholar] [CrossRef]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef]
- Rab, A.; Rowe, S.M.; Raju, S.V.; Bebok, Z.; Matalon, S.; Collawn, J.F. Cigarette smoke and CFTR: Implications in the pathogenesis of COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L530–L541. [Google Scholar] [CrossRef] [PubMed]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Cordts, F.; Pitson, S.; Tabeling, C.; Gibbins, I.; Moffat, D.F.; Jersmann, H.; Hodge, S.; Haberberger, R.V. Expression profile of the sphingosine kinase signalling system in the lung of patients with chronic obstructive pulmonary disease. Life Sci. 2011, 89, 806–811. [Google Scholar] [CrossRef]
- Hamai, H.; Keyserman, F.; Quittell, L.M.; Worgall, T.S. Defective CFTR increases synthesis and mass of sphingolipids that modulate membrane composition and lipid signaling. J. Lipid Res. 2009, 50, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.A.; Meissner, A.; Semenkov, I.; Molinski, S.; Pasyk, S.; Ahmadi, S.; Bui, H.H.; Bear, C.E.; Lidington, D.; Bolz, S.S. Sphingosine-1-Phosphate Is a Novel Regulator of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Activity. PLoS ONE 2015, 10, e0130313. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Don-Doncow, N.; Zhang, Y.; Matuskova, H.; Meissner, A. The emerging alliance of sphingosine-1-phosphate signalling and immune cells: From basic mechanisms to implications in hypertension. Br. J. Pharmacol. 2019, 176, 1989–2001. [Google Scholar] [CrossRef]
- Zamora-Pineda, J.; Kumar, A.; Suh, J.H.; Zhang, M.; Saba, J.D. Dendritic cell sphingosine-1-phosphate lyase regulates thymic egress. J. Exp. Med. 2016, 213, 2773–2791. [Google Scholar] [CrossRef]
- Madej, M.P.; Topfer, E.; Boraschi, D.; Italiani, P. Different Regulation of Interleukin-1 Production and Activity in Monocytes and Macrophages: Innate Memory as an Endogenous Mechanism of IL-1 Inhibition. Front. Pharmacol. 2017, 8, 335. [Google Scholar] [CrossRef]
- Yi, G.; Liang, M.; Li, M.; Fang, X.; Liu, J.; Lai, Y.; Chen, J.; Yao, W.; Feng, X.; Hu, L. A large lung gene expression study identifying IL1B as a novel player in airway inflammation in COPD airway epithelial cells. Inflamm. Res. 2018, 67, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Raleigh, J.M.; Abbate, A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr. Heart Fail. Rep. 2015, 12, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Fuerst, E.; Heiden, D.; Japtok, L.; Diesner, S.C.; Trauner, M.; Kulu, A.; Jaksch, P.; Hoetzenecker, K.; Kleuser, B.; et al. Plasma Levels of the Bioactive Sphingolipid Metabolite S1P in Adult Cystic Fibrosis Patients: Potential Target for Immunonutrition? Nutrients 2020, 12, 765. [Google Scholar] [CrossRef]
- Sattler, K.; Levkau, B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc. Res. 2009, 82, 201–211. [Google Scholar] [CrossRef]
- Hui, S.; Levy, A.S.; Slack, D.L.; Burnstein, M.J.; Errett, L.; Bonneau, D.; Latter, D.; Rotstein, O.D.; Bolz, S.S.; Lidington, D.; et al. Sphingosine-1-Phosphate Signaling Regulates Myogenic Responsiveness in Human Resistance Arteries. PLoS ONE 2015, 10, e0138142. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Serban, K.A.; Schweitzer, K.S.; Bronova, I.A.; Mikosz, A.; Petrache, I. Ceramide and sphingosine-1 phosphate in COPD lungs. Thorax 2021. [Google Scholar] [CrossRef]
- Fernandez Fernandez, E.; De Santi, C.; De Rose, V.; Greene, C.M. CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2018, 12, 483–492. [Google Scholar] [CrossRef]
- Hassan, F.; Xu, X.; Nuovo, G.; Killilea, D.W.; Tyrrell, J.; Da Tan, C.; Tarran, R.; Diaz, P.; Jee, J.; Knoell, D.; et al. Accumulation of metals in GOLD4 COPD lungs is associated with decreased CFTR levels. Respir. Res. 2014, 15, 69. [Google Scholar] [CrossRef]
- Xu, X.; Balsiger, R.; Tyrrell, J.; Boyaka, P.N.; Tarran, R.; Cormet-Boyaka, E. Cigarette smoke exposure reveals a novel role for the MEK/ERK1/2 MAPK pathway in regulation of CFTR. Biochim. Biophys. Acta 2015, 1850, 1224–1232. [Google Scholar] [CrossRef]
- Veltman, M.; Stolarczyk, M.; Radzioch, D.; Wojewodka, G.; De Sanctis, J.B.; Dik, W.A.; Dzyubachyk, O.; Oravecz, T.; de Kleer, I.; Scholte, B.J. Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine-1-phosphate lyase inhibitor LX2931. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1000–L1014. [Google Scholar] [CrossRef]
- Zulueta, A.; Dei Cas, M.; Luciano, F.; Mingione, A.; Pivari, F.; Righi, I.; Morlacchi, L.; Rosso, L.; Signorelli, P.; Ghidoni, R.; et al. Spns2 Transporter Contributes to the Accumulation of S1P in Cystic Fibrosis Human Bronchial Epithelial Cells. Biomedicines 2021, 9, 1121. [Google Scholar] [CrossRef] [PubMed]
- Ammit, A.J.; Hastie, A.T.; Edsall, L.C.; Hoffman, R.K.; Amrani, Y.; Krymskaya, V.P.; Kane, S.A.; Peters, S.P.; Penn, R.B.; Spiegel, S.; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001, 15, 1212–1214. [Google Scholar] [CrossRef]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Huang, L.S.; Natarajan, V. Sphingolipids in pulmonary fibrosis. Adv. Biol. Regul. 2015, 57, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Gorshkova, I.; Zhou, T.; Mathew, B.; Jacobson, J.R.; Takekoshi, D.; Bhattacharya, P.; Smith, B.; Aydogan, B.; Weichselbaum, R.R.; Natarajan, V.; et al. Inhibition of serine palmitoyltransferase delays the onset of radiation-induced pulmonary fibrosis through the negative regulation of sphingosine kinase-1 expression. J. Lipid Res. 2012, 53, 1553–1568. [Google Scholar] [CrossRef]
- Schulz, A.; Tummler, B. Non-allergic asthma as a CFTR-related disorder. J. Cyst. Fibros. 2016, 15, 641–644. [Google Scholar] [CrossRef][Green Version]
- Tzetis, M.; Efthymiadou, A.; Strofalis, S.; Psychou, P.; Dimakou, A.; Pouliou, E.; Doudounakis, S.; Kanavakis, E. CFTR gene mutations—Including three novel nucleotide substitutions—And haplotype background in patients with asthma, disseminated bronchiectasis and chronic obstructive pulmonary disease. Hum. Genet. 2001, 108, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Hodges, C.A.; Cotton, C.U.; Drumm, M.L. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J. Leukoc. Biol. 2012, 92, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Besancon, F.; Przewlocki, G.; Baro, I.; Hongre, A.S.; Escande, D.; Edelman, A. Interferon-gamma downregulates CFTR gene expression in epithelial cells. Am. J. Physiol. 1994, 267, C1398–C1404. [Google Scholar] [CrossRef]
- Ramachandran, S.; Karp, P.H.; Osterhaus, S.R.; Jiang, P.; Wohlford-Lenane, C.; Lennox, K.A.; Jacobi, A.M.; Praekh, K.; Rose, S.D.; Behlke, M.A.; et al. Post-transcriptional regulation of cystic fibrosis transmembrane conductance regulator expression and function by microRNAs. Am. J. Respir. Cell Mol. Biol. 2013, 49, 544–551. [Google Scholar] [CrossRef]
- Raju, S.V.; Jackson, P.L.; Courville, C.A.; McNicholas, C.M.; Sloane, P.A.; Sabbatini, G.; Tidwell, S.; Tang, L.P.; Liu, B.; Fortenberry, J.A.; et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am. J. Respir. Crit. Care Med. 2013, 188, 1321–1330. [Google Scholar] [CrossRef]
- Marklew, A.J.; Patel, W.; Moore, P.J.; Tan, C.D.; Smith, A.J.; Sassano, M.F.; Gray, M.A.; Tarran, R. Cigarette Smoke Exposure Induces Retrograde Trafficking of CFTR to the Endoplasmic Reticulum. Sci. Rep. 2019, 9, 13655. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Pinto, B.F.; Medeiros, N.I.; Teixeira-Carvalho, A.; Eloi-Santos, S.M.; Fontes-Cal, T.C.M.; Rocha, D.A.; Dutra, W.O.; Correa-Oliveira, R.; Gomes, J.A.S. CD86 Expression by Monocytes Influences an Immunomodulatory Profile in Asymptomatic Patients with Chronic Chagas Disease. Front. Immunol. 2018, 9, 454. [Google Scholar] [CrossRef]
- Xu, Y.; Krause, A.; Limberis, M.; Worgall, T.S.; Worgall, S. Low sphingosine-1-phosphate impairs lung dendritic cells in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 48, 250–257. [Google Scholar] [CrossRef]
- Thuy, A.V.; Reimann, C.M.; Hemdan, N.Y.; Graler, M.H. Sphingosine 1-phosphate in blood: Function, metabolism, and fate. Cell. Physiol. Biochem. 2014, 34, 158–171. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kawasaki-Nishi, S.; Otsuka, M.; Hisano, Y.; Yamaguchi, A.; Nishi, T. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 2018, 8, 4969. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.M.; Ishizu, A.N.; Foo, J.C.; Toh, X.R.; Zhang, F.; Whee, D.M.; Torta, F.; Cazenave-Gassiot, A.; Matsumura, T.; Kim, S.; et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017, 550, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Lange, T.; Jungmann, P.; Haberle, J.; Falk, S.; Duebbers, A.; Bruns, R.; Ebner, A.; Hinterdorfer, P.; Oberleithner, H.; Schillers, H. Reduced number of CFTR molecules in erythrocyte plasma membrane of cystic fibrosis patients. Mol. Membr. Biol. 2006, 23, 317–323. [Google Scholar] [CrossRef]
- Ortiz-Munoz, G.; Yu, M.A.; Lefrancais, E.; Mallavia, B.; Valet, C.; Tian, J.J.; Ranucci, S.; Wang, K.M.; Liu, Z.; Kwaan, N.; et al. Cystic fibrosis transmembrane conductance regulator dysfunction in platelets drives lung hyperinflammation. J. Clin. Investig. 2020, 130, 2041–2053. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012, 7, e38941. [Google Scholar] [CrossRef]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Gorshkova, I.; Usatyuk, P.; Kalari, S.; Zhao, Y.; Pyne, N.J.; Pyne, S.; Sabbadini, R.A.; Garcia, J.G.; Natarajan, V. Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: Role of sphingosine kinase 1 and S1P lyase. PLoS ONE 2011, 6, e16571. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef]
- Strong, T.V.; Boehm, K.; Collins, F.S. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J. Clin. Investig. 1994, 93, 347–354. [Google Scholar] [CrossRef]
- Jiang, X.; Shao, Y.; Araj, F.G.; Amin, A.A.; Greenberg, B.M.; Drazner, M.H.; Xing, C.; Mammen, P.P.A. Heterozygous Cystic Fibrosis Transmembrane Regulator Gene Missense Variants Are Associated with Worse Cardiac Function in Patients with Duchenne Muscular Dystrophy. J. Am. Heart Assoc. 2020, 9, e016799. [Google Scholar] [CrossRef]
- Solomon, G.M.; Hathorne, H.; Liu, B.; Raju, S.V.; Reeves, G.; Acosta, E.P.; Dransfield, M.T.; Rowe, S.M. Pilot evaluation of ivacaftor for chronic bronchitis. Lancet Respir. Med. 2016, 4, e32–e33. [Google Scholar] [CrossRef]
- Wiltshire, R.; Nelson, V.; Kho, D.T.; Angel, C.E.; O’Carroll, S.J.; Graham, E.S. Regulation of human cerebro-microvascular endothelial baso-lateral adhesion and barrier function by S1P through dual involvement of S1P1 and S1P2 receptors. Sci. Rep. 2016, 6, 19814. [Google Scholar] [CrossRef]
- Brown, M.B.; Hunt, W.R.; Noe, J.E.; Rush, N.I.; Schweitzer, K.S.; Leece, T.C.; Moldobaeva, A.; Wagner, E.M.; Dudek, S.M.; Poirier, C.; et al. Loss of cystic fibrosis transmembrane conductance regulator impairs lung endothelial cell barrier function and increases susceptibility to microvascular damage from cigarette smoke. Pulm. Circ. 2014, 4, 260–268. [Google Scholar] [CrossRef]
- Heiberg, E.; Sjogren, J.; Ugander, M.; Carlsson, M.; Engblom, H.; Arheden, H. Design and validation of Segment-freely available software for cardiovascular image analysis. BMC Med. Imaging 2010, 10, 1. [Google Scholar] [CrossRef]
- Bode, C.; Graler, M.H. Quantification of sphingosine-1-phosphate and related sphingolipids by liquid chromatography coupled to tandem mass spectrometry. Methods Mol. Biol. 2012, 874, 33–44. [Google Scholar] [CrossRef] [PubMed]






| # S1P1+ Splenic Immune Cells | Sham | HF + Vehicle | HF + C18 |
|---|---|---|---|
| CD3+ T-cells | 10.7 × 104 ± 0.61 × 104 | 17.3 ×104 ± 2.78 × 104 * | 2.1 × 104 ± 0.23 × 104 & |
| Ly6Chi monocytes | 1804 ± 291 | 3388 ± 1008 | 434 ± 39 & |
| Neutrophils | 3288 ± 562 | 15,675 ± 4262 * | 3026 ± 697 & |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uhl, F.E.; Vanherle, L.; Matthes, F.; Meissner, A. Therapeutic CFTR Correction Normalizes Systemic and Lung-Specific S1P Level Alterations Associated with Heart Failure. Int. J. Mol. Sci. 2022, 23, 866. https://doi.org/10.3390/ijms23020866
Uhl FE, Vanherle L, Matthes F, Meissner A. Therapeutic CFTR Correction Normalizes Systemic and Lung-Specific S1P Level Alterations Associated with Heart Failure. International Journal of Molecular Sciences. 2022; 23(2):866. https://doi.org/10.3390/ijms23020866
Chicago/Turabian StyleUhl, Franziska E., Lotte Vanherle, Frank Matthes, and Anja Meissner. 2022. "Therapeutic CFTR Correction Normalizes Systemic and Lung-Specific S1P Level Alterations Associated with Heart Failure" International Journal of Molecular Sciences 23, no. 2: 866. https://doi.org/10.3390/ijms23020866
APA StyleUhl, F. E., Vanherle, L., Matthes, F., & Meissner, A. (2022). Therapeutic CFTR Correction Normalizes Systemic and Lung-Specific S1P Level Alterations Associated with Heart Failure. International Journal of Molecular Sciences, 23(2), 866. https://doi.org/10.3390/ijms23020866