
New Light on an Old Story: Breaking Kasha’s Rule in Phosphorescence Mechanism of Organic Boron Compounds and Molecule Design


Abstract
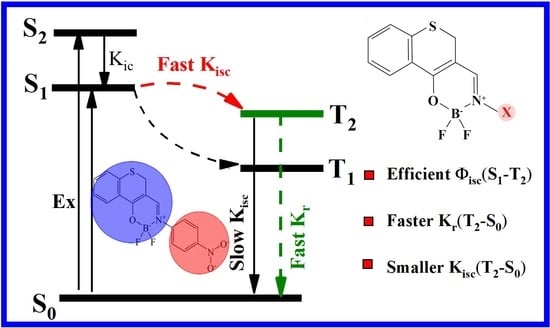
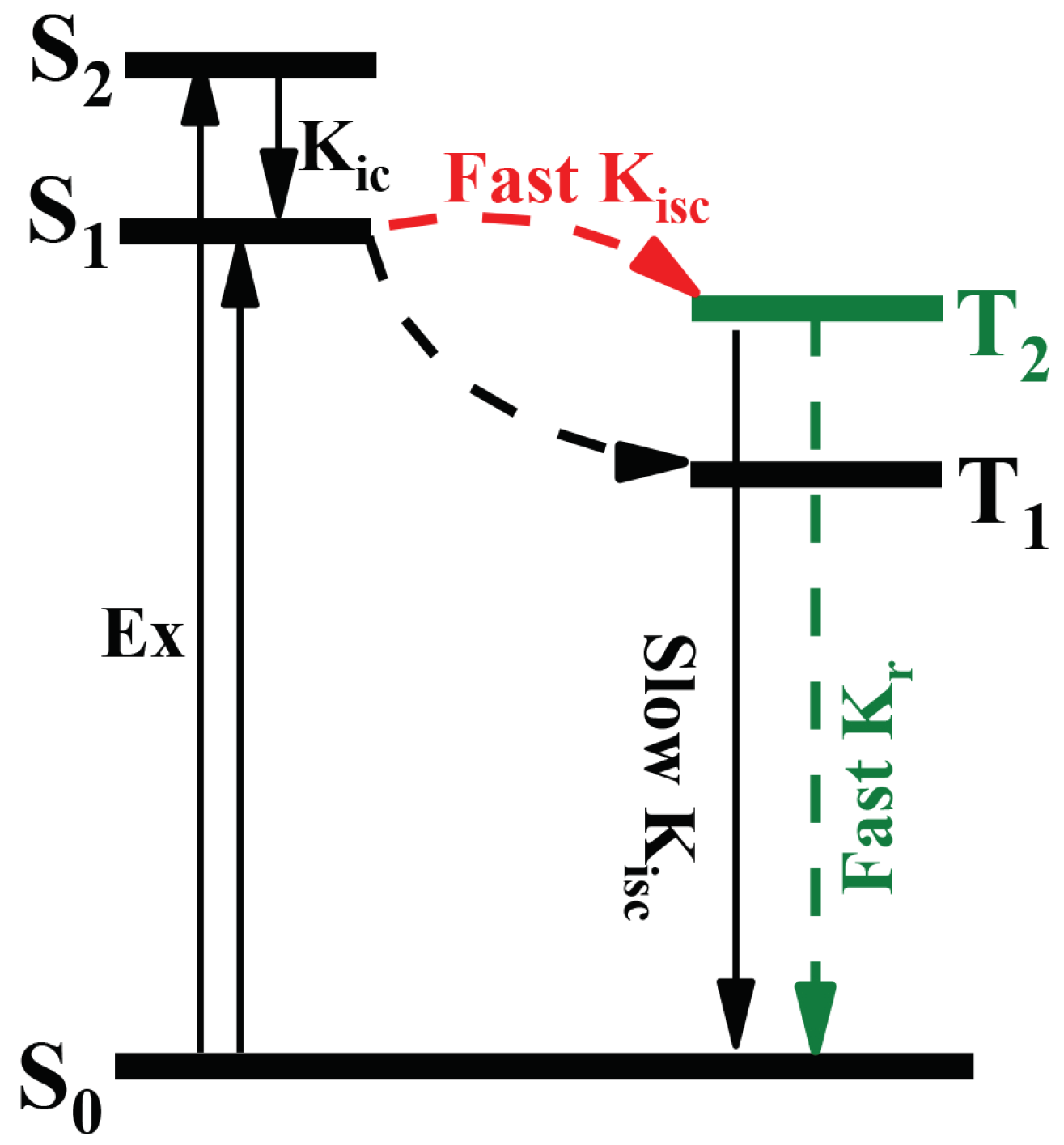
:
1. Introduction
- The vertical emission energy of was in poor agreement with the experimental one.
- To improve the agreement of , Lv et al. tried to use the adiabatic energy rather than the vertical emission one, but only an unusual exchange-correlation (XC) functional could obtain a reasonable result [25].
- Due to Kasha’s rule [26], the probability of (as well as higher triplet states) was not considered at all.
- Paul et al. claimed that TDDFT fails to get multi-reference and [24], which is conditional. In principle, a successful TDDFT calculation requires that (1) the reference state (usually ) by density functional theory (DFT) is single-reference characterized, and (2) the interested excited states may be accessed by one or more single-excitations from . Therefore, an excited state with some multi-reference character may be well calculated by TDDFT only if it is single-excitation dominated from a single-reference (for example, see the third reason on page 4518 of Reference [27]), and of course it is also important to choose suitable XC functionals for some systems like transition metal compounds and charge-transfer (CT) excitations.
- CASSCF and NEVPT2 have a different theoretical basis from TDDFT, and both single-reference and multi-reference states may be described in a unified framework. A crucial question in nearly all the multi-configurational methods is whether adequate static correlations can be captured by the active space, and otherwise multi-configurational calculations merely reproduce single-configurational results or even worse. The NEVPT2 emission wavelength of by Paul et al. is 564 nm [24], at first glance being in good agreement with the experimental phosphorescence peak at 575 nm [23], but it is not clear whether all the important and orbitals have been included in their quite small active space of (4e,4o).
2. Computational Methods
2.1. TDDFT
2.2. ONIOM(DMRG-SCF/MCPDFT:TDDFT)
2.3. Radiative and Non-Radiative Rates
3. Results and Discussion
3.1. Absorption and Emission Energies of S-BF2
3.2. Radiative and Non-radiative Processes of S-BF2
3.3. Newly Designed S-BF2 Derivatives and Their Photoluminescence Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CT | Charge-Transfer |
| DFT | Density Functional Theory |
| DMRG | Density Matrix Renormalization Group |
| IC | Internal Conversion |
| ISC | Intersystem Crossing |
| MCPDFT | Multi-Configurational Pair-Density Functional Theory |
| MECP | Minimum Energy Crossing Point |
| PL | Photoluminescence |
| SOC | Spin-Orbit Coupling |
| TDDFT | Time-Dependent Density Functional Theory |
References
- Chen, H.W.; Lee, J.H.; Lin, B.Y.; Chen, S.; Wu, S.T. Liquid crystal display and organic light-emitting diode display: Present status and future perspectives. Light-Sci. Appl. 2018, 7, 17168. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, Y. Four-coordinate organoboron compounds for organic light-emitting diodes (OLEDs). Chem. Soc. Rev. 2013, 42, 8416–8433. [Google Scholar] [CrossRef]
- Fateminia, S.A.; Mao, Z.; Xu, S.; Yang, Z.; Chi, Z.; Liu, B. Organic nanocrystals with bright red persistent room-temperature phosphorescence for biological applications. Angew. Chem. Int. Ed. 2017, 56, 12160–12164. [Google Scholar] [CrossRef]
- Reineke, S.; Baldo, M.A. Room temperature triplet state spectroscopy of organic semiconductors. Sci. Rep. 2014, 4, 3797. [Google Scholar] [CrossRef] [Green Version]
- Massuyeau, F.; Faulques, E.; Latouche, C. New insights to simulate the luminescence properties of Pt (II) complexes using quantum calculations. J. Chem. Theory Comput. 2017, 13, 1748–1755. [Google Scholar] [CrossRef] [PubMed]
- Younker, J.M.; Dobbs, K.D. Correlating experimental photophysical properties of iridium (III) complexes to spin–orbit coupled TDDFT predictions. J. Phys. Chem. C 2013, 117, 25714–25723. [Google Scholar] [CrossRef]
- Tang, M.C.; Leung, M.Y.; Lai, S.L.; Ng, M.; Chan, M.Y.; Wing-Wah Yam, V. Realization of thermally stimulated delayed phosphorescence in arylgold (III) complexes and efficient gold (III) based blue-emitting organic light-emitting devices. J. Am. Chem. Soc. 2018, 140, 13115–13124. [Google Scholar] [CrossRef] [PubMed]
- Li, E.Y.T.; Jiang, T.Y.; Chi, Y.; Chou, P.T. Semi-quantitative assessment of the intersystem crossing rate: An extension of the El-Sayed rule to the emissive transition metal complexes. Phys. Chem. Chem. Phys. 2014, 16, 26184–26192. [Google Scholar]
- Marian, C.M. Spin–orbit coupling and intersystem crossing in molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Baryshnikov, G.; Minaev, B.; Ågren, H. Theory and calculation of the phosphorescence phenomenon. Chem. Rev. 2017, 117, 6500–6537. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thilagar, P. Recent advances in purely organic phosphorescent materials. Chem. Commun. 2015, 51, 10988–11003. [Google Scholar] [CrossRef]
- Zhao, W.; He, Z.; Lam, J.W.; Peng, Q.; Ma, H.; Shuai, Z.; Bai, G.; Hao, J.; Tang, B.Z. Rational molecular design for achieving persistent and efficient pure organic room-temperature phosphorescence. Chem 2016, 1, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Fu, H. Enhanced Room-Temperature Phosphorescence through Intermolecular Halogen/Hydrogen Bonding. Chem–Eur. J. 2019, 25, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.; Kuno, S. Intersystem crossing mechanisms in the room temperature phosphorescence of crystalline organic compounds. Bull. Chem. Soc. Jpn. 2018, 91, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Hirata, S. Intrinsic analysis of radiative and room-temperature nonradiative processes based on triplet state intramolecular vibrations of heavy atom-free conjugated molecules toward efficient persistent room-temperature phosphorescence. J. Phys. Chem. Lett. 2018, 9, 4251–4259. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhao, W.; Lam, J.W.; Peng, Q.; Ma, H.; Liang, G.; Shuai, Z.; Tang, B.Z. White light emission from a single organic molecule with dual phosphorescence at room temperature. Nat. Commun. 2017, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Peng, Q.; An, Z.; Huang, W.; Shuai, Z. Efficient and long-lived room-temperature organic phosphorescence: Theoretical descriptors for molecular designs. J. Am. Chem. Soc. 2018, 141, 1010–1015. [Google Scholar] [CrossRef]
- Pfister, A.; Zhang, G.; Zareno, J.; Horwitz, A.F.; Fraser, C.L. Boron polylactide nanoparticles exhibiting fluorescence and phosphorescence in aqueous medium. ACS Nano 2008, 2, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Evans, R.E.; Campbell, K.A.; Fraser, C.L. Role of boron in the polymer chemistry and photophysical properties of difluoroboron- dibenzoylmethane polylactide. Macromolecules 2009, 42, 8627–8633. [Google Scholar] [CrossRef]
- Frath, D.; Azizi, S.; Ulrich, G.; Ziessel, R. Chemistry on Boranils: An entry to functionalized fluorescent dyes. Org. Lett. 2012, 14, 4774–4777. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wiltshire, B.D.; Choi, P.; Savin, A.; Sun, S.; Mohammadpour, A.; Ferguson, M.J.; McDonald, R.; Farsinezhad, S.; Brown, A.; et al. Phosphorescence within benzotellurophenes and color tunable tellurophenes under ambient conditions. Chem. Commun. 2015, 51, 5444–5447. [Google Scholar] [CrossRef]
- Torres Delgado, W.; Braun, C.A.; Boone, M.P.; Shynkaruk, O.; Qi, Y.; McDonald, R.; Ferguson, M.J.; Data, P.; Almeida, S.K.; de Aguiar, I.; et al. Moving Beyond Boron-Based Substituents To Achieve Phosphorescence in Tellurophenes. ACS. Appl. Mater. Inter. 2017, 10, 12124–12134. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Wu, Y.; Xiao, L.; Chen, J.; Liao, Q.; Yao, J.; Fu, H. Organic phosphorescence nanowire lasers. J. Am. Chem. Soc. 2017, 139, 6376–6381. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.; Banerjee, A.; Paul, A.; Ruud, K.; Chakrabarti, S. Unraveling the Microscopic Origin of Triplet Lasing from Organic Solids. J. Phys. Chem. Lett. 2018, 9, 4314–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, A.; Ye, W.; Jiang, X.; Gan, N.; Shi, H.; Yao, W.; Ma, H.; An, Z.; Huang, W. Room-Temperature Phosphorescence from Metal-Free Organic Materials in Solution: Origin and Molecular Design. J. Phys. Chem. Lett. 2019, 10, 1037–1042. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Hait, D.; Head-Gordon, M. Orbital Optimized Density Functional Theory for Electronic Excited States. J. Phys. Chem. Lett. 2021, 12, 4517–4529. [Google Scholar] [CrossRef]
- van Gisbergen, S.J.A.; Snijders, J.G.; Baerends, E.J. Implementation of time-dependent density functional response equations. Comput. Phys. Commun. 1999, 118, 119–138. [Google Scholar] [CrossRef]
- Wong, M.W.; Gill, P.M.; Nobes, R.H.; Radom, L. 6-311G (MC)(d, p): A second-row analogue of the 6-311G (d, p) basis set: Calculated heats of formation for second-row hydrides. J. Phys. Chem. 1988, 92, 4875–4880. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phy. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phy. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ruzsinszky, A.; Perdew, J. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.; Chai, J.D. SCAN-based hybrid and double-hybrid density functionals from models without fitted parameters. J. Chem. Phys. 2016, 144, 044114. [Google Scholar] [CrossRef]
- Mezei, P.D.; Csonka, G.I.; Kállay, M. Simple modifications of the SCAN meta-generalized gradient approximation functional. J. Chem. Theory Comput. 2018, 14, 2469–2479. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Liu, W.; Hong, G.; Dai, D.; Li, L.; Dolg, M. The Beijing four-component density functional program package (BDF) and its application to EuO, EuS, YbO and YbS. Theor. Chem. Acc. 1997, 96, 75–83. [Google Scholar] [CrossRef]
- Li, Z.; Suo, B.; Zhang, Y.; Xiao, Y.; Liu, W. Combining spin-adapted open-shell TD-DFT with spin–orbit coupling. Mol. Phys. 2013, 111, 3741–3755. [Google Scholar] [CrossRef]
- Zhang, Y.; Suo, B.; Wang, Z.; Zhang, N.; Li, Z.; Lei, Y.; Zou, W.; Gao, J.; Peng, D.; Pu, Z.; et al. BDF: A relativistic electronic structure program package. J. Chem. Phys. 2020, 152, 064113. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, M.; Sasakura, Y.; Zheng, G.; Morokuma, K. Case Studies of ONIOM(DFT:DFTB) and ONIOM(DFT:DFTB:MM) for Enzymes and Enzyme Mimics. J. Chem. Theory Comput. 2010, 6, 1413–1427. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Aquilante, F.; Pedersen, T.B.; Lindh, R. Low-cost evaluation of the exchange Fock matrix from Cholesky and density fitting representations of the electron repulsion integrals. J. Chem. Phys. 2007, 126, 194106. [Google Scholar] [CrossRef]
- Sharma, P.; Bernales, V.; Knecht, S.; Truhlar, D.G.; Gagliardi, L. Density matrix renormalization group pair-density functional theory (DMRG-PDFT): Singlet-triplet gaps in polyacenes and polyacetylenes. Chem. Sci. 2019, 10, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer, C.E.; Ghosh, S.; Truhlar, D.G.; Gagliardi, L. Multiconfiguration pair-density functional theory is as accurate as CASPT2 for electronic excitation. J. Phys. Chem. Lett. 2016, 7, 586–591. [Google Scholar] [CrossRef]
- Ghosh, S.; Cramer, C.J.; Truhlar, D.G.; Gagliardi, L. Generalized-active-space pair-density functional theory: An efficient method to study large, strongly correlated, conjugated systems. Chem. Sci. 2017, 8, 2741–2750. [Google Scholar] [CrossRef] [Green Version]
- Carlson, R.K.; Truhlar, D.G.; Gagliardi, L. Multiconfiguration pair-density functional theory: A fully translated gradient approximation and its performance for transition metal dimers and the spectroscopy of Re2. J. Chem. Theory Comput. 2015, 11, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Wilbraham, L.; Verma, P.; Truhlar, D.G.; Gagliardi, L.; Ciofini, I. Multiconfiguration pair-density functional theory predicts spin-state ordering in iron complexes with the same accuracy as complete active space second-order perturbation theory at a significantly reduced computational cost. J. Phys. Chem. Lett. 2017, 8, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Adeyiga, O.; Suleiman, O.; Dandu, N.K.; Odoh, S.O. Ground-state actinide chemistry with scalar-relativistic multiconfiguration pair-density functional theory. J. Chem. Phys. 2019, 151, 134102. [Google Scholar] [CrossRef] [PubMed]
- Galván, I.F.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From source code to insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Baiardi, A.; Battaglia, S.; Borin, V.A.; Chibotaru, L.F.; Conti, I.; De Vico, L.; Delcey, M.; Fdez Galván, I.; et al. Modern quantum chemistry with [Open] Molcas. J. Chem. Phys. 2020, 152, 214117. [Google Scholar] [CrossRef]
- Chan, G.K.L.; Sharma, S. The density matrix renormalization group in quantum chemistry. Annu. Rev. Phys. Chem. 2011, 62, 465–481. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Chan, G.K.L. Spin-adapted density matrix renormalization group algorithms for quantum chemistry. J. Chem. Phys. 2012, 136, 124121. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.N.; Aschi, M. Spin-forbidden dehydrogenation of methoxy cation: A statistical view. Phys. Chem. Chem. Phys. 1999, 1, 5555–5563. [Google Scholar] [CrossRef]
- Lu, T. The sobMECP Program. Available online: http://sobereva.com/286 (accessed on 7 January 2022).
- Niu, Y.; Li, W.; Peng, Q.; Geng, H.; Yi, Y.; Wang, L.; Nan, G.; Wang, D.; Shuai, Z. MOlecular MAterials Property Prediction Package (MOMAP) 1.0: A software package for predicting the luminescent properties and mobility of organic functional materials. Mol. Phys. 2018, 116, 1078–1090. [Google Scholar] [CrossRef]
- Itoh, T. Fluorescence and phosphorescence from higher excited states of organic molecules. Chem. Rev. 2012, 112, 4541–4568. [Google Scholar] [CrossRef] [PubMed]
- Mok, D.K.W.; Neumann, R.; Handy, N.C. Dynamical and nondynamical correlation. J. Phys. Chem. 1996, 100, 6225–6230. [Google Scholar] [CrossRef]
- Ghosh, S.; Sonnenberger, A.L.; Hoyer, C.E.; Truhlar, D.G.; Laura, G. Multiconfiguration pair-density functional theory outperforms Kohn-Sham density functional theory and multireference perturbation theory for ground-state and excited-state charge transfer. J. Chem. Theory Comput. 2015, 11, 3643–3649. [Google Scholar] [CrossRef] [PubMed]
- Presti, D.; Truhlar, D.G.; Gagliardi, L. Intramolecular charge transfer and local excitation in organic fluorescent photoredox catalysts explained by RASCI-PDFT. J. Phys. Chem. C 2018, 122, 12061–12070. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functional for spectroscopy: No long-range self-interaction error, good performance for Rydberg and charge-transfer states, and better performance on average than B3LYP for ground states. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef] [PubMed]
- Le Bahers, T.; Adamo, C.; Ciofini, I. A qualitative index of spatial extent in charge-transfer excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Tomin, V.I.; Chou, P.T. Breaking the Kasha rule for more efficient photochemistry. Chem. Rev. 2017, 117, 13353–13381. [Google Scholar] [CrossRef]
- Eber, G.; Grüneis, F.; Schneider, S.; Dörr, F. Dual Fluorescence Emission of Azulene Derivatives in Solution. Chem. Phys. Lett. 1974, 29, 397–404. [Google Scholar] [CrossRef]
- Paul, L.; Chakrabarti, S.; Ruud, K. Anomalous phosphorescence from an organometallic white-light phosphor. J. Phys. Chem. Lett. 2017, 8, 4893–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, L.; Moitra, T.; Ruud, K.; Chakrabarti, S. Strong Duschinsky mixing induced breakdown of Kasha’s rule in an organic phosphor. J. Phys. Chem. Lett. 2019, 10, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.; Li, S.; Fu, L.; Xiao, X.; Xu, Z.; Liao, Q.; Wu, Y.; Yao, J.; Fu, H. Breaking Kasha’s rule as a mechanism for solution-phase room-temperature phosphorescence from high-lying triplet excited state. J. Phys. Chem. Lett. 2020, 11, 8246–8251. [Google Scholar] [CrossRef] [PubMed]
- Englman, R.; Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
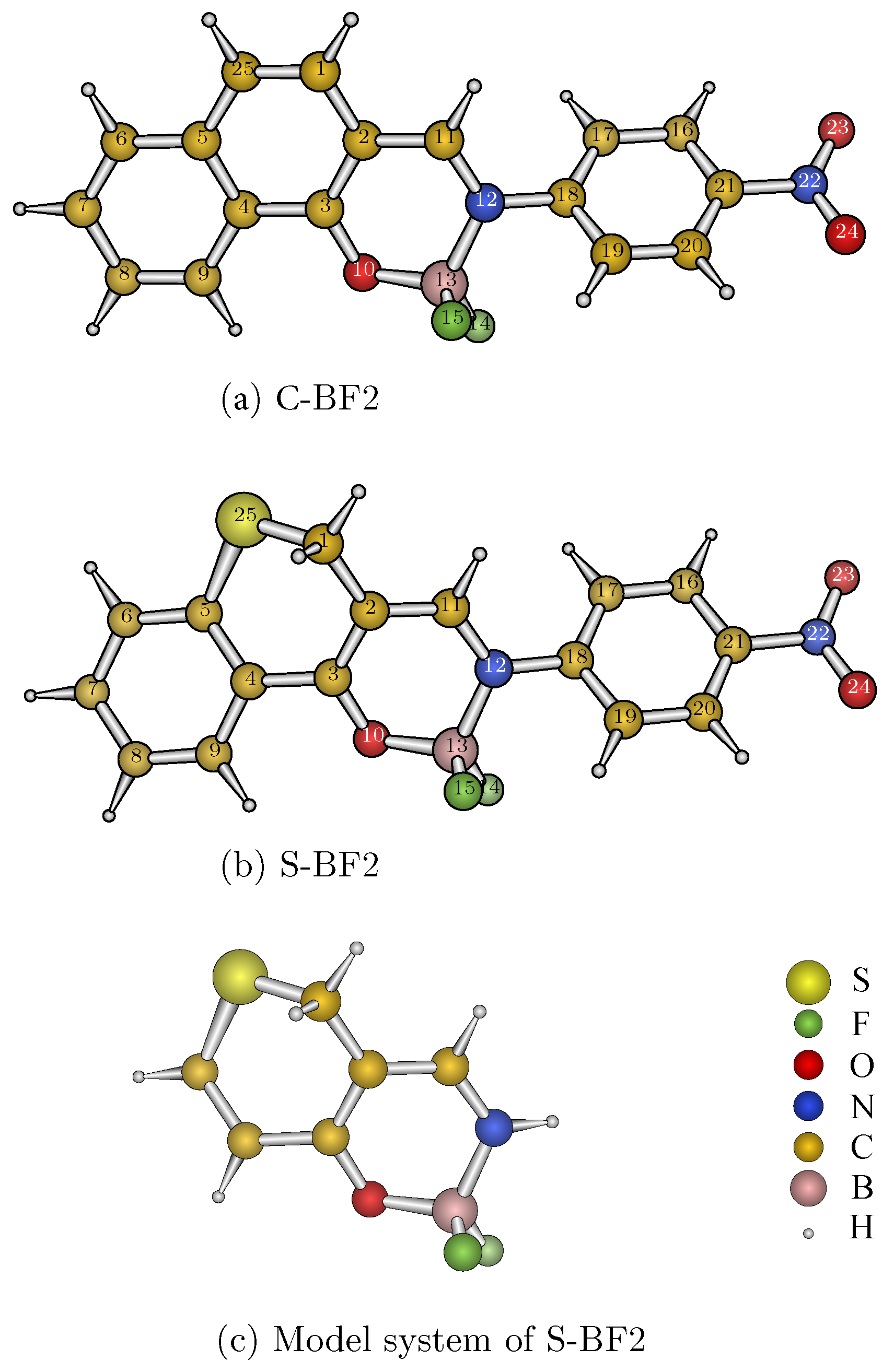



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | ||||
|---|---|---|---|---|
| Model | A | 1.75 (708) | 2.40 (516) | |
| B | 1.74 (714) | 2.38 (520) | ||
| C | 1.77 (702) | 2.42 (512) | ||
| Real (S-BF2) | A | a | 1.87 (664) | 2.07 (598) |
| A | b | 1.93 (643) | 2.16 (574) | |
| B | a | 1.85 (669) | 2.06 (603) | |
| B | b | 1.92 (647) | 2.14 (579) | |
| C | a | 1.88 (659) | 2.10 (592) | |
| C | b | 1.94 (638) | 2.18 (569) | |
| Expt. | 2.16 (575) | |||
| State | E | f | Configuration (%) | |
|---|---|---|---|---|
| Absorption Spectrum | ||||
| 2.85 | 435 | 0.269 | H→L (93) | |
| Expt. | 2.82 | 440 | ||
| 3.30 | 375 | 0.632 | H-1→L (97) | |
| Expt. | 3.26 | 380 | ||
| Phosphorescence Spectrum | ||||
| 1.64 | 758 | 1.31 × 10 | H→L (84), H-1→L (11) | |
| 2.22 | 559 | 1.63 × 10 | H-1→L (53), H→L (38) | |
| Expt. | 2.16 | 575 | ||
| Rate | ||||||
|---|---|---|---|---|---|---|
| 2.80 × 10 | 5.05 × 10 | |||||
| 1.10 × 10 | 5.12 × 10 | 2.24 × 10 | 1.33 × 10 | |||
| 7.37 × 10 | 1.20 × 10 | 3.25 × 10 |
| Molecule | State | E (eV) | (nm) | f | (s) | Configuration (%) |
|---|---|---|---|---|---|---|
| S-BF2* | 1.44 | 864 | 1.65 × 10 | 1.20 × 10 | H→L (95) | |
| 2.36 | 526 | 2.00 × 10 | 4.40 × 10 | H-1→L (78), H→L(15) | ||
| S-BF2_C | 1.18 | 1050 | 6.74 × 10 | 3.42 × 10 | H→L (94) | |
| 2.34 | 530 | 4.05 × 10 | 8.76 × 10 | H-1→L (89) | ||
| S-BF2_N | 1.21 | 1025 | 6.82 × 10 | 3.66 × 10 | H→L (92) | |
| 2.35 | 527 | 5.47 × 10 | 1.19 × 10 | H-2→L (89) | ||
| S-BF2_O | 1.33 | 931 | 7.10 × 10 | 4.57 × 10 | H→L (92) | |
| 2.33 | 533 | 1.25 × 10 | 2.68 × 10 | H-1→L (90) | ||
| S-BF2_S | 1.35 | 917 | 7.44 × 10 | 4.71 × 10 | H→L (92) | |
| 2.33 | 533 | 8.31 × 10 | 1.77 × 10 | H-2→L (89) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, D.; Suo, B.; Zou, W. New Light on an Old Story: Breaking Kasha’s Rule in Phosphorescence Mechanism of Organic Boron Compounds and Molecule Design. Int. J. Mol. Sci. 2022, 23, 876. https://doi.org/10.3390/ijms23020876
Deng D, Suo B, Zou W. New Light on an Old Story: Breaking Kasha’s Rule in Phosphorescence Mechanism of Organic Boron Compounds and Molecule Design. International Journal of Molecular Sciences. 2022; 23(2):876. https://doi.org/10.3390/ijms23020876
Chicago/Turabian StyleDeng, Dan, Bingbing Suo, and Wenli Zou. 2022. "New Light on an Old Story: Breaking Kasha’s Rule in Phosphorescence Mechanism of Organic Boron Compounds and Molecule Design" International Journal of Molecular Sciences 23, no. 2: 876. https://doi.org/10.3390/ijms23020876
APA StyleDeng, D., Suo, B., & Zou, W. (2022). New Light on an Old Story: Breaking Kasha’s Rule in Phosphorescence Mechanism of Organic Boron Compounds and Molecule Design. International Journal of Molecular Sciences, 23(2), 876. https://doi.org/10.3390/ijms23020876