
Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells


Abstract
:1. Introduction
2. Epidemiology
3. Diagnosis
4. Molecular Mechanisms in the Pathogenesis
4.1. Synovium
4.2. T Cell-Mediated Immune Response in RA
4.3. B Cell-Mediated Immune Response in RA
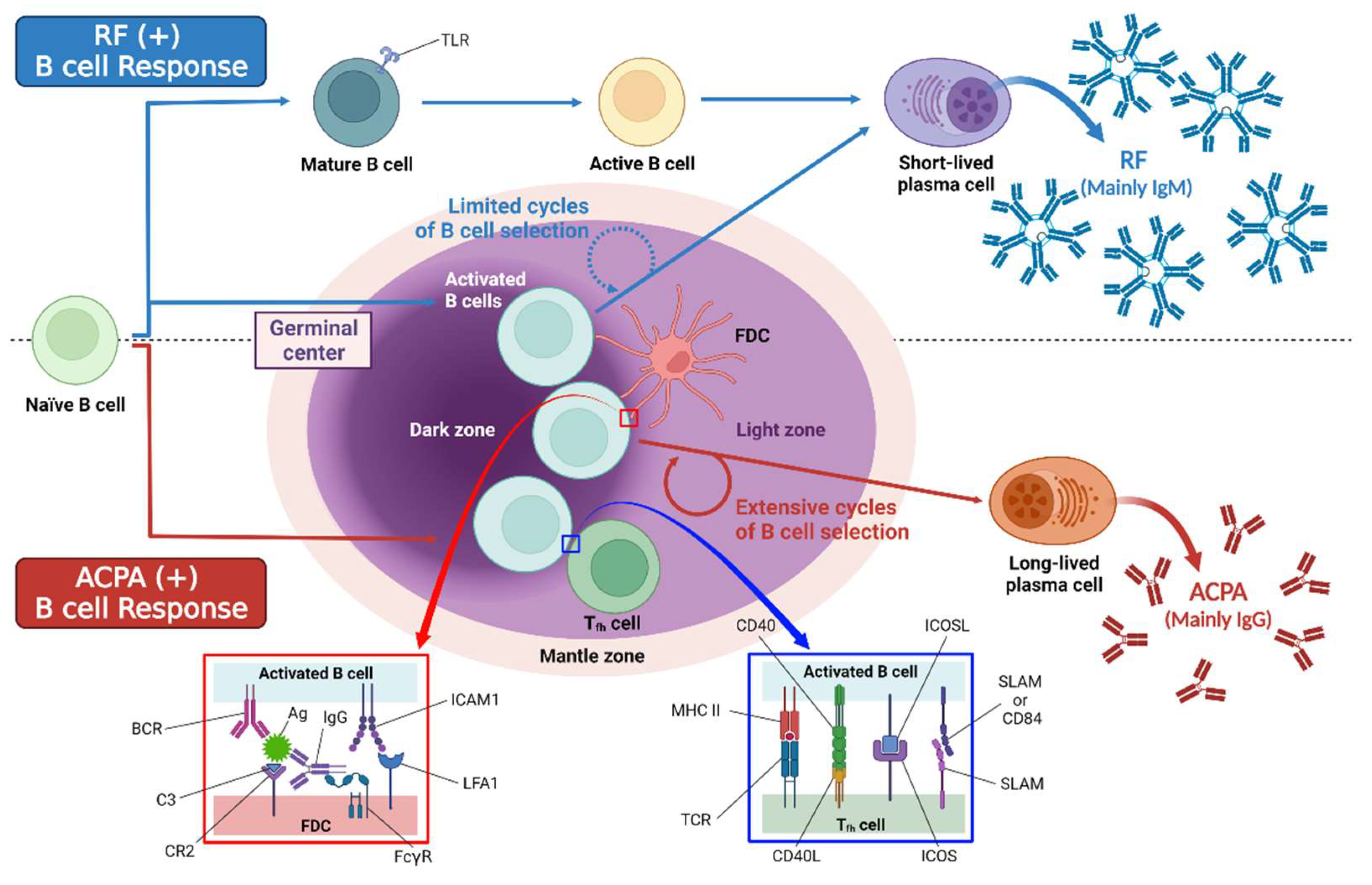
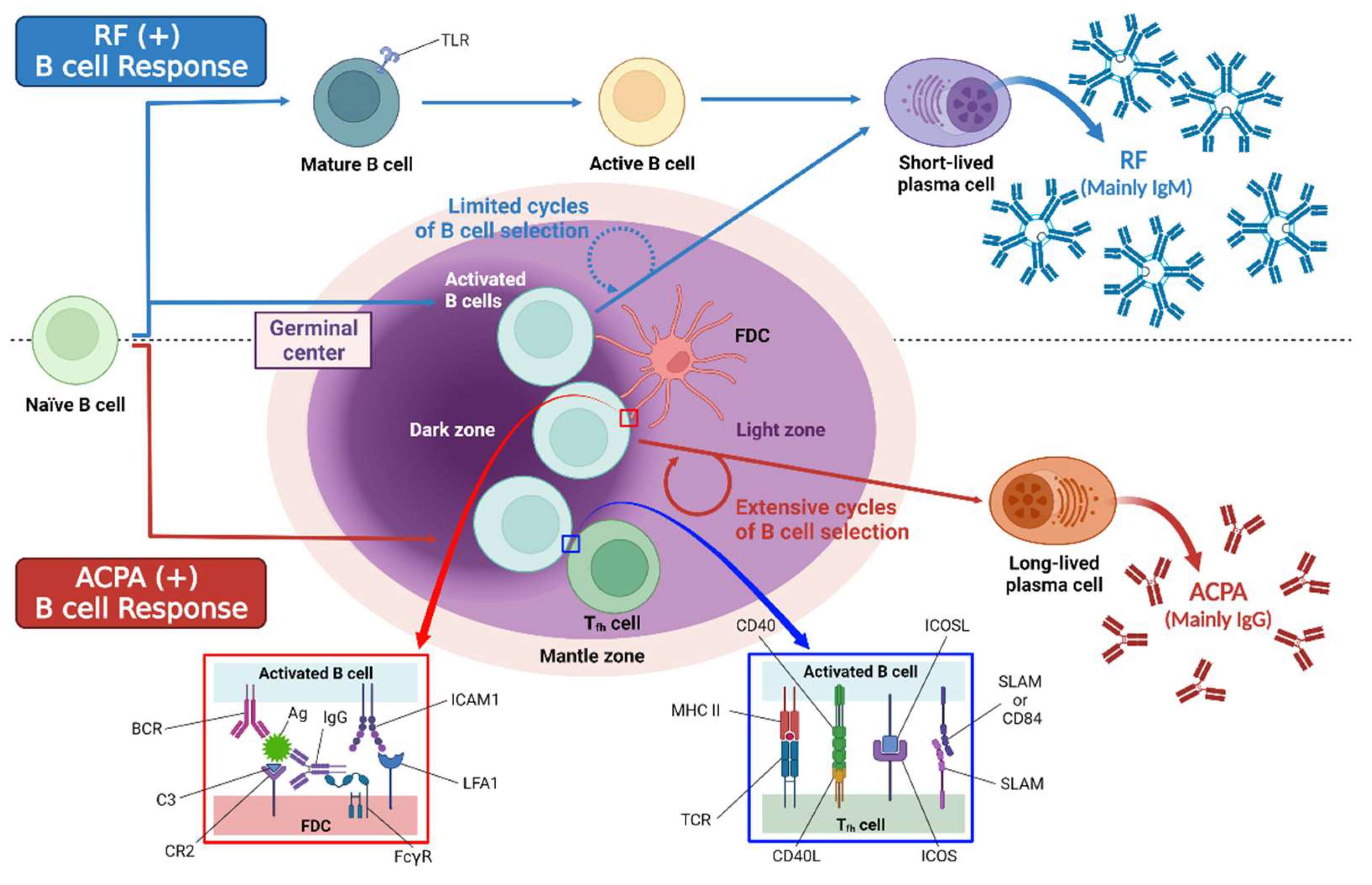
4.3.1. Autoantibody Production
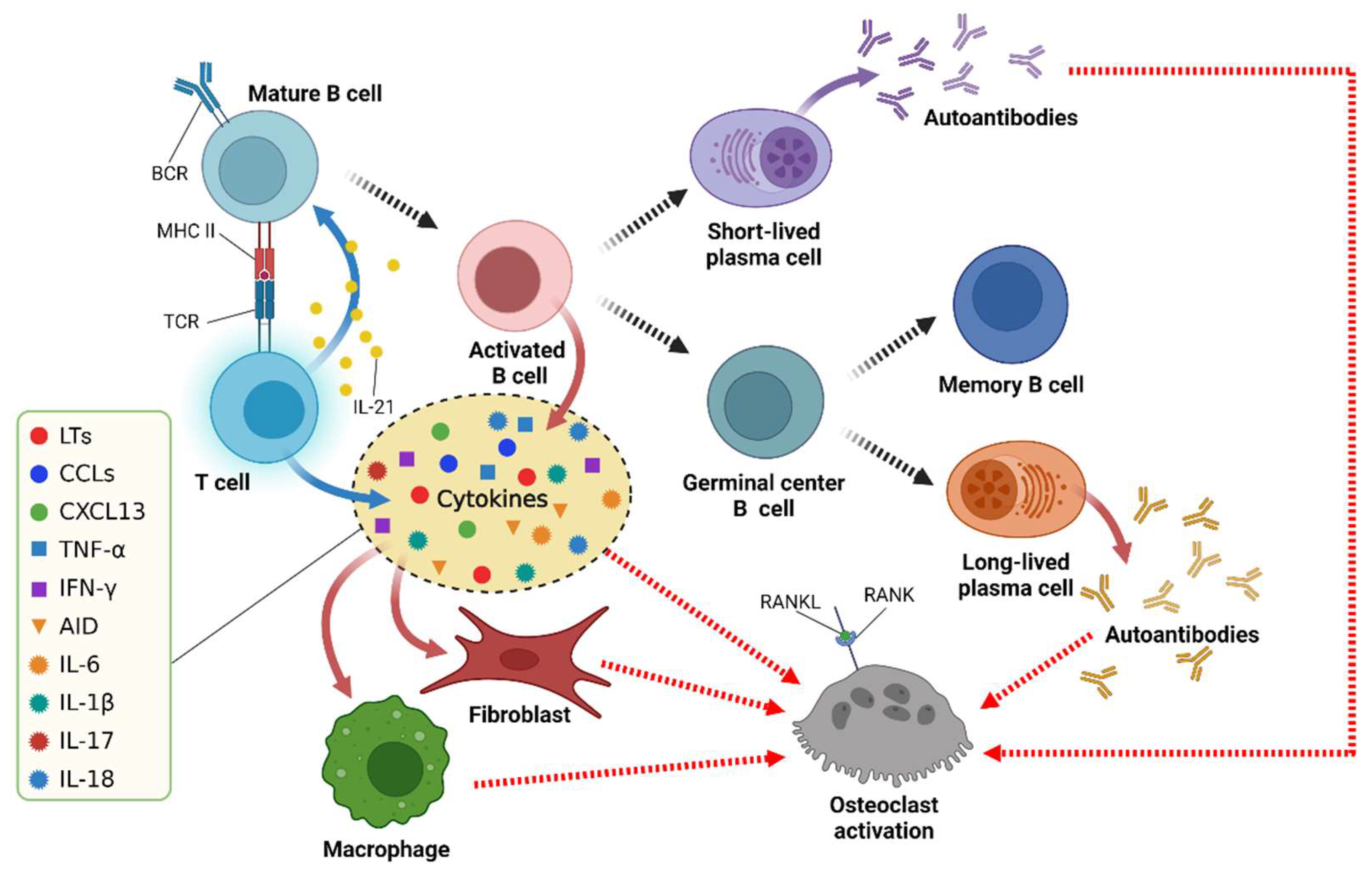
4.3.2. Antigen Presentation
4.3.3. Cytokine Secretion
4.3.4. Osteoclast Activation
4.4. Innate Immunity-Mediated Immune Response in RA
5. Treatments Targeting the Pathogenic Cells and Cytokines
5.1. Currently Approved Biologic/Targeted Syntetic DMARDS
5.2. Therapeutic Approaches under Evaluation in Humans
5.3. Potential Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doan, T.; Massarotti, E. Rheumatoid arthritis: An overview of new and emerging therapies. J. Clin. Pharmacol. 2005, 45, 751–762. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheumatol. 2010, 62, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowson, C.S.; Matteson, E.L.; Myasoedova, E.; Michet, C.J.; Ernste, F.C.; Warrington, K.J.; Davis, J.M., 3rd; Hunder, G.G.; Therneau, T.M.; Gabriel, S.E. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheumatol. 2011, 63, 633–639. [Google Scholar] [CrossRef] [Green Version]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheumatol. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Consortium, W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Raychaudhuri, S.; Thomson, B.P.; Remmers, E.F.; Eyre, S.; Hinks, A.; Guiducci, C.; Catanese, J.J.; Xie, G.; Stahl, E.A.; Chen, R.; et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat. Genet. 2009, 41, 1313–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Thomson, W.; Barton, A.; Ke, X.; Eyre, S.; Hinks, A.; Bowes, J.; Donn, R.; Symmons, D.; Hider, S.; Bruce, I.N.; et al. Rheumatoid arthritis association at 6q23. Nat. Genet. 2007, 39, 1431–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, A.; Thomson, W.; Ke, X.; Eyre, S.; Hinks, A.; Bowes, J.; Plant, D.; Gibbons, L.J.; Wilson, A.G.; Bax, D.E.; et al. Rheumatoid arthritis susceptibility loci at chromosomes 10p15, 12q13 and 22q13. Nat. Genet. 2008, 40, 1156–1159. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheumatol. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishimura, K.; Tamaki, K.; Tsuji, G.; Nakazawa, T.; Morinobu, A.; Kumagai, S. Impact of smoking as a risk factor for developing rheumatoid arthritis: A meta-analysis of observational studies. Ann. Rheum. Dis. 2010, 69, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolt, P.; Källberg, H.; Lundberg, I.; Sjögren, B.; Klareskog, L.; Alfredsson, L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: Results from the Swedish EIRA study. Ann. Rheum. Dis. 2005, 64, 582–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Kay, J.; Upchurch, K.S. ACR/EULAR 2010 rheumatoid arthritis classification criteria. Rheumatology 2012, 51 (Suppl. 6), vi5–vi9. [Google Scholar] [CrossRef] [Green Version]
- Casey, J. Anti-MCV antibodies—A promising new biomarker for RA. Nat. Rev. Rheumatol. 2009, 5, 179. [Google Scholar] [CrossRef]
- Zhu, J.N.; Nie, L.Y.; Lu, X.Y.; Wu, H.X. Meta-analysis: Compared with anti-CCP and rheumatoid factor, could anti-MCV be the next biomarker in the rheumatoid arthritis classification criteria? Clin. Chem. Lab. Med. 2019, 57, 1668–1679. [Google Scholar] [CrossRef]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. B Cells in Rheumatoid Arthritis: Pathogenic Mechanisms and Treatment Prospects. Front. Immunol. 2021, 12, 750753. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Ouboussad, L.; Burska, A.N.; Melville, A.; Buch, M.H. Synovial Tissue Heterogeneity in Rheumatoid Arthritis and Changes With Biologic and Targeted Synthetic Therapies to Inform Stratified Therapy. Front. Med. 2019, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromley, M.; Woolley, D.E. Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheumatol. 1984, 27, 857–863. [Google Scholar] [CrossRef]
- Firestein, G.S. Etiology and pathogenesis of rheumatoid arthritis. In Kelley’s Textbook of Rheumatology; Elsevier: Amsterdam, The Netherlands, 2001; pp. 921–966. [Google Scholar]
- Scherer, H.; Kerkman, P.; van der Voort, E.; Trouw, L.; Huizinga, T.; Toes, R. ACPA production by circulating B cells isolated from peripheral blood of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71 (Suppl. 1), A33. [Google Scholar] [CrossRef] [Green Version]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.K.; Quinn, J.M.; Sims, N.A.; van Nieuwenhuijze, A.; Campbell, I.K.; Wicks, I.P. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheumatol. 2006, 54, 158–168. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Autoimmune arthritis: The interface between the immune system and joints. Adv. Immunol. 2012, 115, 45–71. [Google Scholar]
- Wu, X.; Tian, J.; Wang, S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 1112. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, P.; Zhou, Y.; Meng, L.; Zhu, W.; Jiang, C.; Wang, L.; Cai, Y.; Lu, S.; Hou, W. Increased expression of Th17 cytokines and interleukin-22 correlates with disease activity in pristane-induced arthritis in rats. PLoS ONE 2017, 12, e0188199. [Google Scholar] [CrossRef] [Green Version]
- Lotfi, N.; Thome, R.; Rezaei, N.; Zhang, G.X.; Rezaei, A.; Rostami, A.; Esmaeil, N. Roles of GM-CSF in the Pathogenesis of Autoimmune Diseases: An Update. Front. Immunol. 2019, 10, 1265. [Google Scholar] [CrossRef]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Gaffen, S.L. Structure-function relationships in the IL-17 receptor: Implications for signal transduction and therapy. Cytokine 2008, 41, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthys, P.; Vermeire, K.; Mitera, T.; Heremans, H.; Huang, S.; Billiau, A. Anti-IL-12 antibody prevents the development and progression of collagen-induced arthritis in IFN-gamma receptor-deficient mice. Eur. J. Immunol. 1998, 28, 2143–2151. [Google Scholar] [CrossRef]
- Nakae, S.; Nambu, A.; Sudo, K.; Iwakura, Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 2003, 171, 6173–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, H. Targeting interleukin-17 in patients with active rheumatoid arthritis: Rationale and clinical potential. Ther. Adv. Musculoskelet. Dis. 2013, 5, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Blanco, F.J.; Möricke, R.; Dokoupilova, E.; Codding, C.; Neal, J.; Andersson, M.; Rohrer, S.; Richards, H. Secukinumab in Active Rheumatoid Arthritis: A Phase III Randomized, Double-Blind, Active Comparator- and Placebo-Controlled Study. Arthritis Rheumatol. 2017, 69, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Ikeuchi, H.; Kuroiwa, T.; Hiramatsu, N.; Kaneko, Y.; Hiromura, K.; Ueki, K.; Nojima, Y. Expression of interleukin-22 in rheumatoid arthritis: Potential role as a proinflammatory cytokine. Arthritis Rheumatol. 2005, 52, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, E.J.; Prakken, B.J.; van Wijk, F. T cells out of control—Impaired immune regulation in the inflamed joint. Nat. Rev. Rheumatol. 2013, 9, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Koenen, H.J.; Smeets, R.L.; Vink, P.M.; van Rijssen, E.; Boots, A.M.; Joosten, I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [Google Scholar] [CrossRef]
- Li, X.; Zheng, Y. Regulatory T cell identity: Formation and maintenance. Trends Immunol. 2015, 36, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Hibino, S.; Someya, K.; Yoshmura, A. Regulation of regulatory T cells: Epigenetics and plasticity. Adv. Immunol. 2014, 124, 249–273. [Google Scholar]
- Barbi, J.; Pardoll, D.; Pan, F. Treg functional stability and its responsiveness to the microenvironment. Immunol. Rev. 2014, 259, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Panayi, G.S. Even though T-cell-directed trials have been of limited success, is there reason for optimism? Nat. Clin. Pract. Rheumatol. 2006, 2, 58–59. [Google Scholar] [CrossRef]
- Alenazy, M.F.; Saheb Sharif-Askari, F.; Omair, M.A.; El-Wetidy, M.S.; Omair, M.A.; Mitwalli, H.; Al-Muhsen, S.; Al-Masri, A.; Hamid, Q.; Halwani, R. Abatacept enhances blood regulatory B cells of rheumatoid arthritis patients to a level that associates with disease remittance. Sci. Rep. 2021, 11, 5629. [Google Scholar] [CrossRef]
- Bugatti, S.; Vitolo, B.; Caporali, R.; Montecucco, C.; Manzo, A. B cells in rheumatoid arthritis: From pathogenic players to disease biomarkers. BioMed Res. Int. 2014, 2014, 681678. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.S.; Verheul, M.K.; Stoop, J.N.; Liu, B.; Ioan-Facsinay, A.; van Veelen, P.A.; de Ru, A.H.; Janssen, G.M.C.; Hegen, M.; Rapecki, S.; et al. Breach of autoreactive B cell tolerance by post-translationally modified proteins. Ann. Rheum. Dis. 2017, 76, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hsu, H.C.; Chen, J.; Grizzle, W.E.; Chatham, W.W.; Stockard, C.R.; Wu, Q.; Yang, P.A.; Holers, V.M.; Mountz, J.D. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand. J. Immunol. 2009, 70, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, C.G.; Sanz, I.; Cook, M.C. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009, 9, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Turesson, C.; Kastbom, A. Autoantibodies in Rheumatoid Arthritis—Laboratory and Clinical Perspectives. Front. Immunol. 2021, 12, 685312. [Google Scholar] [CrossRef] [PubMed]
- Malmström, V.; Grönwall, C. The parallel worlds of ACPA-positive and RF-positive B cells. Nat. Rev. Rheumatol. 2018, 14, 626–628. [Google Scholar] [CrossRef]
- van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef]
- Lotze, M.T.; Thomson, A.W. Measuring Immunity: Basic Biology and Clinical Assessment; Elsevier Academic Press: Amsterdam, The Netherlands, 2005; pp. 187–192. [Google Scholar]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid factors: Clinical applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Dörner, T.; Egerer, K.; Feist, E.; Burmester, G.R. Rheumatoid factor revisited. Curr. Opin. Rheumatol. 2004, 16, 246–253. [Google Scholar] [CrossRef]
- Barra, L.; Bykerk, V.; Pope, J.E.; Haraoui, B.P.; Hitchon, C.A.; Thorne, J.C.; Keystone, E.C.; Boire, G. Anticitrullinated protein antibodies and rheumatoid factor fluctuate in early inflammatory arthritis and do not predict clinical outcomes. J. Rheumatol. 2013, 40, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Volkov, M.; van Schie, K.A.; van der Woude, D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol. Rev. 2020, 294, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Pruijn, G.J.; Wiik, A.; van Venrooij, W.J. The use of citrullinated peptides and proteins for the diagnosis of rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, 203. [Google Scholar] [CrossRef] [Green Version]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheumatol. 2004, 50, 380–386. [Google Scholar] [CrossRef]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheumatol. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Kwon, E.J.; Ju, J.H. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. Int. J. Mol. Sci. 2021, 22, 10576. [Google Scholar] [CrossRef]
- Masson-Bessière, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nogueira, L.; Vincent, C.; Senshu, T.; Serre, G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J. Immunol. 2001, 166, 4177–4184. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, H.; Fisher, B.A.; Källberg, H.; Plant, D.; Malmström, V.; Rönnelid, J.; Charles, P.; Ding, B.; Alfredsson, L.; Padyukov, L.; et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 2009, 41, 1319–1324. [Google Scholar] [CrossRef]
- Verpoort, K.N.; Cheung, K.; Ioan-Facsinay, A.; van der Helm-van Mil, A.H.; de Vries-Bouwstra, J.K.; Allaart, C.F.; Drijfhout, J.W.; de Vries, R.R.; Breedveld, F.C.; Huizinga, T.W.; et al. Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheumatol. 2007, 56, 3949–3952. [Google Scholar] [CrossRef]
- Burkhardt, H.; Sehnert, B.; Bockermann, R.; Engström, A.; Kalden, J.R.; Holmdahl, R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur. J. Immunol. 2005, 35, 1643–1652. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Kulik, L.; Tomooka, B.; Braschler, K.J.; Arend, W.P.; Robinson, W.H.; Holers, V.M. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J. Clin. Investig. 2006, 116, 961–973. [Google Scholar] [CrossRef]
- Kurowska, W.; Kuca-Warnawin, E.H.; Radzikowska, A.; Maśliński, W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent. Eur. J. Immunol. 2017, 42, 390–398. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheumatol. 2008, 58, 678–688. [Google Scholar] [CrossRef]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.; Toes, R.E. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheumatol. 2009, 60, 1923–1931. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheumatol. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Anquetil, F.; Clavel, C.; Offer, G.; Serre, G.; Sebbag, M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J. Immunol. 2015, 194, 3664–3674. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Chen, X.; Jensen, P.E. The role of B lymphocytes as antigen-presenting cells. Arch. Immunol. Ther. Exp. 2008, 56, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Perdriger, A.; Amé, P. Definition of B cell helper T cells in rheumatoid arthritis and their behavior during treatment. Semin. Arthritis Rheum. 2020, 50, 867–872. [Google Scholar] [CrossRef]
- van den Berg, W.B.; McInnes, I.B. Th17 cells and IL-17 a—Focus on immunopathogenesis and immunotherapeutics. Semin. Arthritis Rheum. 2013, 43, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Chi, S.; Wang, X.; Sun, J.; Zhang, Y. CD4+CXCR5+PD-1+ T Follicular Helper Cells Play a Pivotal Role in the Development of Rheumatoid Arthritis. Med. Sci. Monit. 2019, 25, 3032–3040. [Google Scholar] [CrossRef]
- Yanaba, K.; Bouaziz, J.D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Li, F.; Li, X.; Wang, Z.G.; Zhang, B. TNF-α and RANKL promote osteoclastogenesis by upregulating RANK via the NF-κB pathway. Mol. Med. Rep. 2018, 17, 6605–6611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, B.; Jiang, Z.; Jiang, Y. Reduced numbers of regulatory B cells are negatively correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Zhang, L.; Chen, J.; Zhu, M.; Hou, L.; Chen, B.; Shen, B. Changes in regulatory B cells and their relationship with rheumatoid arthritis disease activity. Clin. Exp. Med. 2015, 15, 285–292. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci. Transl. Med. 2013, 5, 173ra23. [Google Scholar] [CrossRef]
- Marston, B.; Palanichamy, A.; Anolik, J.H. B cells in the pathogenesis and treatment of rheumatoid arthritis. Curr. Opin. Rheumatol. 2010, 22, 307–315. [Google Scholar] [CrossRef]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef]
- Mizukami, J.; Takaesu, G.; Akatsuka, H.; Sakurai, H.; Ninomiya-Tsuji, J.; Matsumoto, K.; Sakurai, N. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol. Cell. Biol. 2002, 22, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocijan, R.; Finzel, S.; Englbrecht, M.; Engelke, K.; Rech, J.; Schett, G. Differences in bone structure between rheumatoid arthritis and psoriatic arthritis patients relative to autoantibody positivity. Ann. Rheum. Dis. 2014, 73, 2022–2028. [Google Scholar] [CrossRef]
- Komatsu, N.; Win, S.; Yan, M.; Huynh, N.C.; Sawa, S.; Tsukasaki, M.; Terashima, A.; Pluemsakunthai, W.; Kollias, G.; Nakashima, T.; et al. Plasma cells promote osteoclastogenesis and periarticular bone loss in autoimmune arthritis. J. Clin. Investig. 2021, 131, e143060. [Google Scholar] [CrossRef]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed. J. 2021, 44, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Ardura, J.A.; Rackov, G.; Izquierdo, E.; Alonso, V.; Gortazar, A.R.; Escribese, M.M. Targeting Macrophages: Friends or Foes in Disease? Front. Pharmacol. 2019, 10, 1255. [Google Scholar] [CrossRef]
- Kinne, R.W.; Stuhlmüller, B.; Burmester, G.R. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res. Ther. 2007, 9, 224. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, S.; Iwamoto, N.; Takatani, A.; Igawa, T.; Shimizu, T.; Umeda, M.; Nishino, A.; Horai, Y.; Hirai, Y.; Koga, T.; et al. M1 and M2 Monocytes in Rheumatoid Arthritis: A Contribution of Imbalance of M1/M2 Monocytes to Osteoclastogenesis. Front. Immunol. 2017, 8, 1958. [Google Scholar] [CrossRef] [Green Version]
- Iberg, C.A.; Jones, A.; Hawiger, D. Dendritic Cells As Inducers of Peripheral Tolerance. Trends Immunol. 2017, 38, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.B.; Langridge, W.H.R. The function of myeloid dendritic cells in rheumatoid arthritis. Rheumatol. Int. 2017, 37, 1043–1051. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Gundle, R.; Davies, R.J.; Lee, Y.C.; McMichael, A.J.; Callan, M.F. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J. Immunol. 2004, 173, 6418–6426. [Google Scholar] [CrossRef] [Green Version]
- Tak, P.P.; Kummer, J.A.; Hack, C.E.; Daha, M.R.; Smeets, T.J.; Erkelens, G.W.; Meinders, A.E.; Kluin, P.M.; Breedveld, F.C. Granzyme-positive cytotoxic cells are specifically increased in early rheumatoid synovial tissue. Arthritis Rheumatol. 1994, 37, 1735–1743. [Google Scholar] [CrossRef]
- Rodríguez-Carrio, J.; Hähnlein, J.S.; Ramwadhdoebe, T.H.; Semmelink, J.F.; Choi, I.Y.; van Lienden, K.P.; Maas, M.; Gerlag, D.M.; Tak, P.P.; Geijtenbeek, T.B.; et al. Brief Report: Altered Innate Lymphoid Cell Subsets in Human Lymph Node Biopsy Specimens Obtained During the At-Risk and Earliest Phases of Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 70–76. [Google Scholar] [CrossRef]
- Takaki-Kuwahara, A.; Arinobu, Y.; Miyawaki, K.; Yamada, H.; Tsuzuki, H.; Irino, K.; Ayano, M.; Kimoto, Y.; Mitoma, H.; Akahoshi, M.; et al. CCR6+ group 3 innate lymphoid cells accumulate in inflamed joints in rheumatoid arthritis and produce Th17 cytokines. Arthritis Res. Ther. 2019, 21, 198. [Google Scholar] [CrossRef] [Green Version]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamri, F.; de Vries, T.J. Use of TNF Inhibitors in Rheumatoid Arthritis and Implications for the Periodontal Status: For the Benefit of Both? Front. Immunol. 2020, 11, 591365. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2013, 1, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2013, 8, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Vital, E.M.; Emery, P. Abatacept in the treatment of rheumatoid arthritis. Ther. Clin. Risk Manag. 2006, 2, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, M.G.; Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Profile of sarilumab and its potential in the treatment of rheumatoid arthritis. Drug Des. Dev. Ther. 2017, 11, 1593–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef]
- Kubo, S.; Nakayamada, S.; Tanaka, Y. Baricitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2016, 12, 911–919. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J. Inflamm. Res. 2020, 13, 519–531. [Google Scholar] [CrossRef]
- Cole, S.; Walsh, A.; Yin, X.; Wechalekar, M.D.; Smith, M.D.; Proudman, S.M.; Veale, D.J.; Fearon, U.; Pitzalis, C.; Humby, F.; et al. Integrative analysis reveals CD38 as a therapeutic target for plasma cell-rich pre-disease and established rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 85. [Google Scholar] [CrossRef]
- Good, L.; Benner, B.; Carson, W.E. Bruton’s tyrosine kinase: An emerging targeted therapy in myeloid cells within the tumor microenvironment. Cancer Immunol. Immunother. 2021, 70, 2439–2451. [Google Scholar] [CrossRef] [PubMed]
- Arneson, L.C.; Carroll, K.J.; Ruderman, E.M. Bruton’s Tyrosine Kinase Inhibition for the Treatment of Rheumatoid Arthritis. Immunotargets Ther. 2021, 10, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.L.; Hu, W.; Lu, W.G.; Li, X.; Li, J.P.; Xu, R.S.; Li, C.W.; Chen, F.H.; Jin, C. Targeting interleukin-21 in rheumatoid arthritis. Mol. Biol. Rep. 2011, 38, 1717–1721. [Google Scholar] [CrossRef]
- Ignatenko, S.; Skrumsager, B.K.; Mouritzen, U. Safety, PK, and PD of recombinant anti-interleukin-21 monoclonal antibody in a first-in-human trial. Int. J. Clin. Pharmacol. Ther. 2016, 54, 243–252. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.M.; Greenwald, M.; Satterwhite, J.; Veenhuizen, M.; Xie, L.; Berclaz, P.Y.; Myers, S.; Benichou, O. Tabalumab, an anti-BAFF monoclonal antibody, in patients with active rheumatoid arthritis with an inadequate response to TNF inhibitors. Ann. Rheum. Dis. 2013, 72, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Smolen, J.S.; Weinblatt, M.E.; van der Heijde, D.; Rigby, W.F.; van Vollenhoven, R.; Bingham, C.O., 3rd; Veenhuizen, M.; Gill, A.; Zhao, F.; Komocsar, W.J.; et al. Efficacy and safety of tabalumab, an anti-B-cell-activating factor monoclonal antibody, in patients with rheumatoid arthritis who had an inadequate response to methotrexate therapy: Results from a phase III multicentre, randomised, double-blind study. Ann. Rheum. Dis. 2015, 74, 1567–1570. [Google Scholar] [CrossRef]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann. Rheum. Dis. 2021, 80, 176–184. [Google Scholar] [CrossRef]
- Gowhari Shabgah, A.; Shariati-Sarabi, Z.; Tavakkol-Afshari, J.; Ghasemi, A.; Ghoryani, M.; Mohammadi, M. A significant decrease of BAFF, APRIL, and BAFF receptors following mesenchymal stem cell transplantation in patients with refractory rheumatoid arthritis. Gene 2020, 732, 144336. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshabrawy, H.A.; Essani, A.E.; Szekanecz, Z.; Fox, D.A.; Shahrara, S. TLRs, future potential therapeutic targets for RA. Autoimmun. Rev. 2017, 16, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Classification Criteria for RA (Total Score ≥ 6 is Considered Satisfactory for the Diagnosis of RA) | Score | |
|---|---|---|
| joint involvement (swollen or tender joint) | 1 large joint (shoulders, elbows, hips, knees, and ankles) | 0 |
| 2–10 large joints | 1 | |
| 1–3 small joints (with or without involvement of large joints) * | 2 | |
| 4–10 small joints (with or without involvement of large joints) | 3 | |
| >10 joints (at least 1 small joint) ** | 5 | |
| serology | Negative RF and negative ACPA (≤upper limit of normal (ULN)) | 0 |
| Low-positive RF or low-positive ACPA (≤ULN and ≤3 times) | 2 | |
| High-positive RF or high-positive ACPA (≤3 times) | 3 | |
| acute-phase reactants | Normal CRP and normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 | |
| duration of symptoms | <6 weeks | 0 |
| ≥6 weeks | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.; Kwon, E.-J.; Lee, J.J. Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. Int. J. Mol. Sci. 2022, 23, 905. https://doi.org/10.3390/ijms23020905
Jang S, Kwon E-J, Lee JJ. Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. International Journal of Molecular Sciences. 2022; 23(2):905. https://doi.org/10.3390/ijms23020905
Chicago/Turabian StyleJang, Sunhee, Eui-Jong Kwon, and Jennifer Jooha Lee. 2022. "Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells" International Journal of Molecular Sciences 23, no. 2: 905. https://doi.org/10.3390/ijms23020905
APA StyleJang, S., Kwon, E. -J., & Lee, J. J. (2022). Rheumatoid Arthritis: Pathogenic Roles of Diverse Immune Cells. International Journal of Molecular Sciences, 23(2), 905. https://doi.org/10.3390/ijms23020905