

Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Amyloid β Peptide
3. Aβ/APP-Cellular Membrane and Lipid Influence
4. Lipid Rafts
5. Cholesterol and Pathogenesis of AD
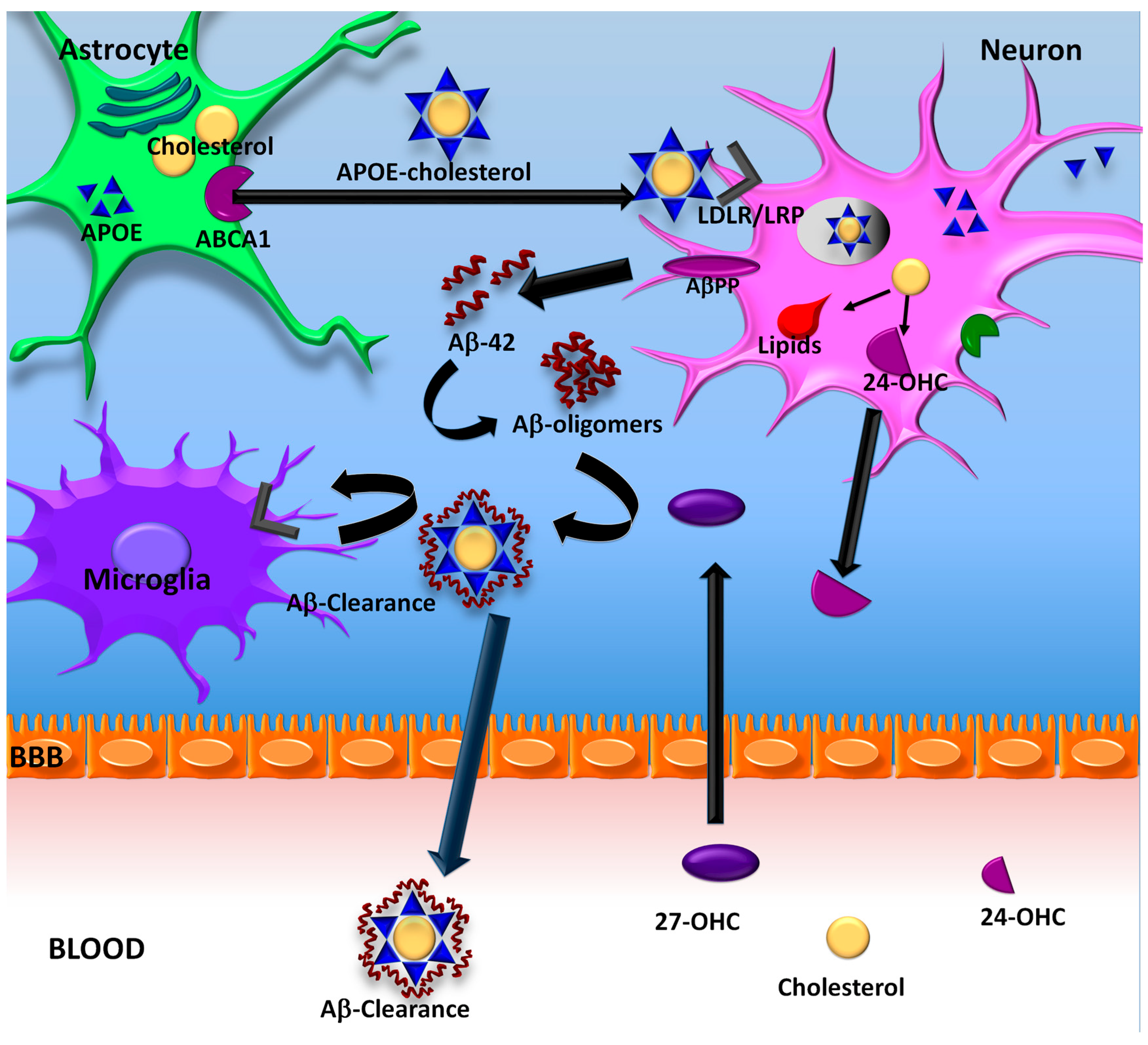
6. ApoE Lipidation
7. ApoE Lipidation and Aβ
ApoE Lipidation as a Therapeutic Target for AD
8. Cholinergic System
Basal Forebrain and Cognition
9. nAChRs, Lipids and Aβ
10. Therapy
10.1. Statins
10.2. AChE Inhibitors
10.2.1. Tacrine (TCR)
10.2.2. Donepezil (DPZ)
10.2.3. Rivastigmine (RVG)
10.2.4. Galantamine (GLT)
10.3. Nanotherapy
10.3.1. Nanodelivery of AChE Inhibitors to Improve Their Effectiveness in AD Treatment
10.3.2. Nanodelivery of Tacrine
10.3.3. Nanodelivery of Donepezil
10.3.4. Nanodelivery of Rivastigmine
10.3.5. Nanodelivery of Galantamine
11. Supramolecular Strategies
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Nakazato, Y.; Kawarabayashi, T.; Ishiguro, K.; Ihara, Y.; Morimatsu, M.; Hirai, S. Extracellular neurofibrillary tangles associated with degenerating neurites and neuropil threads in Alzheimer-type dementia. Acta Neuropathol. 1991, 81, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Terry, R.D.; Mallory, M.; Alford, M.; Hansen, L.A. Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am. J. Pathol. 1990, 137, 1293–1297. [Google Scholar] [PubMed]
- Mandelkow, E.; von Bergen, M.; Biernat, J.; Mandelkow, E.M. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007, 17, 83–90. [Google Scholar] [CrossRef]
- Meraz-Rios, M.A.; Lira-De Leon, K.I.; Campos-Pena, V.; De Anda-Hernandez, M.A.; Mena-Lopez, R. Tau oligomers and aggregation in Alzheimer’s disease. J. Neurochem. 2010, 112, 1353–1367. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Delacourte, A.; Defossez, A. Alzheimer’s disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 1986, 76, 173–186. [Google Scholar] [CrossRef]
- Campos-Pena, V.; Toral-Rios, D.; Becerril-Perez, F.; Sanchez-Torres, C.; Delgado-Namorado, Y.; Torres-Ossorio, E.; Franco-Bocanegra, D.; Carvajal, K. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: Is Abeta a Crucial Factor in Both Pathologies? Antioxid. Redox Signal. 2017, 26, 542–560. [Google Scholar] [CrossRef]
- Dries, D.R.; Yu, G. Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 132–146. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- Iversen, L.L.; Mortishire-Smith, R.J.; Pollack, S.J.; Shearman, M.S. The toxicity in vitro of beta-amyloid protein. Biochem. J. 1995, 311 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Oltersdorf, T.; Teplow, D.B.; Selkoe, D.J. Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann. N. Y. Acad. Sci. 1993, 695, 109–116. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Hartmann, H.; Eckert, A.; Muller, W.E. Apolipoprotein E and cholesterol affect neuronal calcium signalling: The possible relationship to beta-amyloid neurotoxicity. Biochem. Biophys. Res. Commun. 1994, 200, 1185–1192. [Google Scholar] [CrossRef]
- Subasinghe, S.; Unabia, S.; Barrow, C.J.; Mok, S.S.; Aguilar, M.I.; Small, D.H. Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J. Neurochem. 2003, 84, 471–479. [Google Scholar] [CrossRef]
- Barenholz, Y. Sphingomyelin and cholesterol: From membrane biophysics and rafts to potential medical applications. Subcell. Biochem. 2004, 37, 167–215. [Google Scholar]
- Kellner, R.R.; Baier, C.J.; Willig, K.I.; Hell, S.W.; Barrantes, F.J. Nanoscale organization of nicotinic acetylcholine receptors revealed by stimulated emission depletion microscopy. Neuroscience 2007, 144, 135–143. [Google Scholar] [CrossRef]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S. Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Proteolytic processing of Alzheimer’s beta-amyloid precursor protein. J. Neurochem. 2012, 120 (Suppl. S1), 9–21. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef]
- Novo, M.; Freire, S.; Al-Soufi, W. Critical aggregation concentration for the formation of early Amyloid-beta (1-42) oligomers. Sci. Rep. 2018, 8, 1783. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; George, L.; Tung, Y.C.; Kim, K.S.; Wisniewski, H.M. Amyloid protein and neurofibrillary tangles coexist in the same neuron in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 2853–2857. [Google Scholar] [CrossRef]
- Wirths, O.; Walter, S.; Kraus, I.; Klafki, H.W.; Stazi, M.; Oberstein, T.J.; Ghiso, J.; Wiltfang, J.; Bayer, T.A.; Weggen, S. N-truncated Abeta4-x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimer’s Res. Ther. 2017, 9, 80. [Google Scholar] [CrossRef]
- Guzman, E.A.; Bouter, Y.; Richard, B.C.; Lannfelt, L.; Ingelsson, M.; Paetau, A.; Verkkoniemi-Ahola, A.; Wirths, O.; Bayer, T.A. Abundance of Abeta(5)-x like immunoreactivity in transgenic 5XFAD, APP/PS1KI and 3xTG mice, sporadic and familial Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 13. [Google Scholar] [CrossRef]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid beta (Abeta) 4-42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- McKee, A.C.; Kosik, K.S.; Kowall, N.W. Neuritic pathology and dementia in Alzheimer’s disease. Ann. Neurol. 1991, 30, 156–165. [Google Scholar] [CrossRef]
- Nagy, Z.; Esiri, M.M.; Jobst, K.A.; Morris, J.H.; King, E.M.; McDonald, B.; Litchfield, S.; Barnetson, L. Clustering of pathological features in Alzheimer’s disease: Clinical and neuroanatomical aspects. Dementia 1996, 7, 121–127. [Google Scholar]
- Arendt, T.; Bigl, V.; Arendt, A.; Tennstedt, A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol. 1983, 61, 101–108. [Google Scholar] [CrossRef]
- Hanna Al-Shaikh, F.S.; Duara, R.; Crook, J.E.; Lesser, E.R.; Schaeverbeke, J.; Hinkle, K.M.; Ross, O.A.; Ertekin-Taner, N.; Pedraza, O.; Dickson, D.W.; et al. Selective Vulnerability of the Nucleus Basalis of Meynert Among Neuropathologic Subtypes of Alzheimer Disease. JAMA Neurol. 2020, 77, 225–233. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fa, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef]
- Goni, F.M. The basic structure and dynamics of cell membranes: An update of the Singer-Nicolson model. Biochim. Biophys. Acta 2014, 1838, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ashkar, R. The dynamic face of lipid membranes. Soft Matter 2021, 17, 6910–6928. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Yoda, M.; Miura, T.; Takeuchi, H. Non-electrostatic binding and self-association of amyloid beta-peptide on the surface of tightly packed phosphatidylcholine membranes. Biochem. Biophys. Res. Commun. 2008, 376, 56–59. [Google Scholar] [CrossRef]
- Peters, I.; Igbavboa, U.; Schutt, T.; Haidari, S.; Hartig, U.; Rosello, X.; Bottner, S.; Copanaki, E.; Deller, T.; Kogel, D.; et al. The interaction of beta-amyloid protein with cellular membranes stimulates its own production. Biochim. Biophys. Acta 2009, 1788, 964–972. [Google Scholar] [CrossRef]
- Seelig, J. On the flexibility of hydrocarbon chains in lipid bilayers. J. Am. Chem. Soc. 1971, 93, 5017–5022. [Google Scholar] [CrossRef]
- Vanni, S.; Riccardi, L.; Palermo, G.; De Vivo, M. Structure and Dynamics of the Acyl Chains in the Membrane Trafficking and Enzymatic Processing of Lipids. Acc. Chem. Res. 2019, 52, 3087–3096. [Google Scholar] [CrossRef]
- Niu, Z.; Zhang, Z.; Zhao, W.; Yang, J. Interactions between amyloid beta peptide and lipid membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1663–1669. [Google Scholar] [CrossRef]
- Tan, J.Z.A.; Gleeson, P.A. The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 2019, 1861, 697–712. [Google Scholar] [CrossRef]
- Holmes, O.; Paturi, S.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Effects of membrane lipids on the activity and processivity of purified gamma-secretase. Biochemistry 2012, 51, 3565–3575. [Google Scholar] [CrossRef]
- Verdier, Y.; Zarandi, M.; Penke, B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 2004, 10, 229–248. [Google Scholar] [CrossRef]
- Rangachari, V.; Dean, D.N.; Rana, P.; Vaidya, A.; Ghosh, P. Cause and consequence of Abeta—Lipid interactions in Alzheimer disease pathogenesis. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1652–1662. [Google Scholar] [CrossRef]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Abeta(1-40) and Abeta(1-42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef]
- Fatafta, H.; Khaled, M.; Owen, M.C.; Sayyed-Ahmad, A.; Strodel, B. Amyloid-beta peptide dimers undergo a random coil to beta-sheet transition in the aqueous phase but not at the neuronal membrane. Proc. Natl. Acad. Sci. USA 2021, 118, e2106210118. [Google Scholar] [CrossRef]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar]
- Di Scala, C.; Yahi, N.; Lelievre, C.; Garmy, N.; Chahinian, H.; Fantini, J. Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s beta amyloid peptide. ACS Chem. Neurosci. 2013, 4, 509–517. [Google Scholar] [CrossRef]
- Hashemi, M.; Banerjee, S.; Lyubchenko, Y.L. Free Cholesterol Accelerates Abeta Self-Assembly on Membranes at Physiological Concentration. Int. J. Mol. Sci. 2022, 23, 2803. [Google Scholar] [CrossRef]
- Kandel, N.; Matos, J.O.; Tatulian, S.A. Structure of amyloid beta25-35 in lipid environment and cholesterol-dependent membrane pore formation. Sci. Rep. 2019, 9, 2689. [Google Scholar] [CrossRef]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Nickels, J.D.; Smith, M.D.; Alsop, R.J.; Himbert, S.; Yahya, A.; Cordner, D.; Zolnierczuk, P.; Stanley, C.B.; Katsaras, J.; Cheng, X.; et al. Lipid Rafts: Buffers of Cell Membrane Physical Properties. J. Phys. Chem. B 2019, 123, 2050–2056. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lecart, S.; Saudou, F.; Duyckaerts, C.; Leveque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, N.; Takami, M.; Okochi, M.; Wada-Kakuda, S.; Fujiwara, H.; Tagami, S.; Funamoto, S.; Ihara, Y.; Morishima-Kawashima, M. gamma-Secretase associated with lipid rafts: Multiple interactive pathways in the stepwise processing of beta-carboxyl-terminal fragment. J. Biol. Chem. 2014, 289, 5109–5121. [Google Scholar] [CrossRef] [PubMed]
- Mousa, Y.M.; Abdallah, I.M.; Hwang, M.; Martin, D.R.; Kaddoumi, A. Author Correction: Amylin and pramlintide modulate gamma-secretase level and APP processing in lipid rafts. Sci. Rep. 2020, 10, 11096. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef]
- Ikonen, E. Roles of lipid rafts in membrane transport. Curr. Opin. Cell Biol. 2001, 13, 470–477. [Google Scholar] [CrossRef]
- Ariotti, N.; Fernandez-Rojo, M.A.; Zhou, Y.; Hill, M.M.; Rodkey, T.L.; Inder, K.L.; Tanner, L.B.; Wenk, M.R.; Hancock, J.F.; Parton, R.G. Caveolae regulate the nanoscale organization of the plasma membrane to remotely control Ras signaling. J. Cell Biol. 2014, 204, 777–792. [Google Scholar] [CrossRef]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes. J. Lipid Res. 2020, 61, 676–686. [Google Scholar] [CrossRef]
- Santos, A.L.; Preta, G. Lipids in the cell: Organisation regulates function. Cell Mol. Life Sci. 2018, 75, 1909–1927. [Google Scholar] [CrossRef]
- Moll, T.; Marshall, J.N.G.; Soni, N.; Zhang, S.; Cooper-Knock, J.; Shaw, P.J. Membrane lipid raft homeostasis is directly linked to neurodegeneration. Essays Biochem. 2021, 65, 999–1011. [Google Scholar]
- Angelopoulou, E.; Paudel, Y.N.; Shaikh, M.F.; Piperi, C. Flotillin: A Promising Biomarker for Alzheimer’s Disease. J. Pers. Med. 2020, 10, 20. [Google Scholar] [CrossRef]
- Koch, J.C.; Solis, G.P.; Bodrikov, V.; Michel, U.; Haralampieva, D.; Shypitsyna, A.; Tonges, L.; Bahr, M.; Lingor, P.; Stuermer, C.A. Upregulation of reggie-1/flotillin-2 promotes axon regeneration in the rat optic nerve in vivo and neurite growth in vitro. Neurobiol. Dis. 2013, 51, 168–176. [Google Scholar] [CrossRef]
- Genest, M.; Comunale, F.; Planchon, D.; Govindin, P.; Noly, D.; Vacher, S.; Bieche, I.; Robert, B.; Malhotra, H.; Schoenit, A.; et al. Upregulated flotillins and sphingosine kinase 2 derail AXL vesicular traffic to promote epithelial-mesenchymal transition. J. Cell Sci. 2022, 135, jcs259178. [Google Scholar] [CrossRef]
- Neumann-Giesen, C.; Fernow, I.; Amaddii, M.; Tikkanen, R. Role of EGF-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J. Cell Sci. 2007, 120 Pt 3, 395–406. [Google Scholar] [CrossRef]
- Riento, K.; Frick, M.; Schafer, I.; Nichols, B.J. Endocytosis of flotillin-1 and flotillin-2 is regulated by Fyn kinase. J. Cell Sci. 2009, 122 Pt 7, 912–918. [Google Scholar] [CrossRef]
- Lee, S.J.; Liyanage, U.; Bickel, P.E.; Xia, W.; Lansbury, P.T., Jr.; Kosik, K.S. A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 1998, 4, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Oshima, N.; Morishima-Kawashima, M.; Yamaguchi, H.; Yoshimura, M.; Sugihara, S.; Khan, K.; Games, D.; Schenk, D.; Ihara, Y. Accumulation of amyloid beta-protein in the low-density membrane domain accurately reflects the extent of beta-amyloid deposition in the brain. Am. J. Pathol. 2001, 158, 2209–2218. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef]
- Goritz, C.; Mauch, D.H.; Pfrieger, F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell Neurosci. 2005, 29, 190–201. [Google Scholar] [CrossRef]
- Sprong, H.; van der Sluijs, P.; van Meer, G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001, 2, 504–513. [Google Scholar] [CrossRef]
- Thiele, C.; Hannah, M.J.; Fahrenholz, F.; Huttner, W.B. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 2000, 2, 42–49. [Google Scholar] [CrossRef]
- Koudinov, A.R.; Koudinova, N.V. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 2001, 15, 1858–1860. [Google Scholar] [CrossRef]
- Posse de Chaves, E. Reciprocal regulation of cholesterol and beta amyloid at the subcellular level in Alzheimer’s disease. Can. J. Physiol. Pharmacol. 2012, 90, 753–764. [Google Scholar] [CrossRef]
- He, X.Y.; Dobkin, C.; Yang, S.Y. 17beta-Hydroxysteroid dehydrogenases and neurosteroid metabolism in the central nervous system. Mol. Cell Endocrinol. 2019, 489, 92–97. [Google Scholar] [CrossRef]
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Mondal, M. Sterol and lipid trafficking in mammalian cells. Biochem. Soc. Trans. 2006, 34 Pt 3, 335–339. [Google Scholar] [CrossRef]
- Rog, T.; Vattulainen, I. Cholesterol, sphingolipids, and glycolipids: What do we know about their role in raft-like membranes? Chem. Phys. Lipids 2014, 184, 82–104. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yamauchi, Y.; Hasan, M.T.; Chang, C. Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res. 2017, 58, 2239–2254. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef]
- Morell, P.; Jurevics, H. Origin of cholesterol in myelin. Neurochem. Res. 1996, 21, 463–470. [Google Scholar] [CrossRef]
- DeGrella, R.F.; Simoni, R.D. Intracellular transport of cholesterol to the plasma membrane. J. Biol. Chem. 1982, 257, 14256–14262. [Google Scholar] [CrossRef]
- Heino, S.; Lusa, S.; Somerharju, P.; Ehnholm, C.; Olkkonen, V.M.; Ikonen, E. Dissecting the role of the golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc. Natl. Acad. Sci. USA 2000, 97, 8375–8380. [Google Scholar] [CrossRef]
- Vance, J.E.; Pan, D.; Campenot, R.B.; Bussiere, M.; Vance, D.E. Evidence that the major membrane lipids, except cholesterol, are made in axons of cultured rat sympathetic neurons. J. Neurochem. 1994, 62, 329–337. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Brown, M.S.; Li, W.P.; Nohturfft, A.; Goldstein, J.L.; Espenshade, P.J. Transport-dependent proteolysis of SREBP: Relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 1999, 99, 703–712. [Google Scholar] [CrossRef]
- Nohturfft, A.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; Espenshade, P.J. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 2000, 102, 315–323. [Google Scholar] [CrossRef]
- Thelen, K.M.; Falkai, P.; Bayer, T.A.; Lutjohann, D. Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci. Lett. 2006, 403, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Lipid compositions of different regions of the human brain during aging. J. Neurochem. 1990, 54, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [Google Scholar] [CrossRef]
- Chou, Y.C.; Lin, S.B.; Tsai, L.H.; Tsai, H.I.; Lin, C.M. Cholesterol deficiency increases the vulnerability of hippocampal glia in primary culture to glutamate-induced excitotoxicity. Neurochem. Int. 2003, 43, 197–209. [Google Scholar] [CrossRef]
- Brinton, R.D. Neurosteroids as regenerative agents in the brain: Therapeutic implications. Nat. Rev. Endocrinol. 2013, 9, 241–250. [Google Scholar] [CrossRef]
- Kao, Y.C.; Ho, P.C.; Tu, Y.K.; Jou, I.M.; Tsai, K.J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Puglielli, L.; Tanzi, R.E.; Kovacs, D.M. Alzheimer’s disease: The cholesterol connection. Nat. Neurosci. 2003, 6, 345–351. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- De Oliveira, J.; Moreira, E.L.; dos Santos, D.B.; Piermartiri, T.C.; Dutra, R.C.; Pinton, S.; Tasca, C.I.; Farina, M.; Prediger, R.D.; de Bem, A.F. Increased susceptibility to amyloid-beta-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J. Alzheimer’s Dis. 2014, 41, 43–60. [Google Scholar] [CrossRef]
- Ibrahim Fouad, G. Combination of Omega 3 and Coenzyme Q10 Exerts Neuroprotective Potential Against Hypercholesterolemia-Induced Alzheimer’s-Like Disease in Rats. Neurochem. Res. 2020, 45, 1142–1155. [Google Scholar] [CrossRef]
- Moreira, E.L.; de Oliveira, J.; Engel, D.F.; Walz, R.; de Bem, A.F.; Farina, M.; Prediger, R.D. Hypercholesterolemia induces short-term spatial memory impairments in mice: Up-regulation of acetylcholinesterase activity as an early and causal event? J. Neural Transm. 2014, 121, 415–426. [Google Scholar] [CrossRef]
- Moreira, E.L.; de Oliveira, J.; Nunes, J.C.; Santos, D.B.; Nunes, F.C.; Vieira, D.S.; Ribeiro-do-Valle, R.M.; Pamplona, F.A.; de Bem, A.F.; Farina, M.; et al. Age-related cognitive decline in hypercholesterolemic LDL receptor knockout mice (LDLr-/-): Evidence of antioxidant imbalance and increased acetylcholinesterase activity in the prefrontal cortex. J. Alzheimer’s Dis. 2012, 32, 495–511. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef]
- Lanfranco, M.F.; Ng, C.A.; Rebeck, G.W. ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6336. [Google Scholar] [CrossRef]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin. Lipidol. 2010, 51, 555–573. [Google Scholar] [CrossRef]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 2004, 279, 14009–14015. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 13–21. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Reinshagen, V.; Zhou, S.; Burgess, B.L.; Bernier, L.; McIsaac, S.A.; Chan, J.Y.; Tansley, G.H.; Cohn, J.S.; Hayden, M.R.; Wellington, C.L. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 2004, 279, 41197–41207. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Oram, J.F. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J. Lipid Res. 2006, 47, 2433–2443. [Google Scholar] [CrossRef]
- Burgess, B.L.; Parkinson, P.F.; Racke, M.M.; Hirsch-Reinshagen, V.; Fan, J.; Wong, C.; Stukas, S.; Theroux, L.; Chan, J.Y.; Donkin, J.; et al. ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J. Lipid Res. 2008, 49, 1254–1267. [Google Scholar] [CrossRef]
- Ji, Z.S.; Pitas, R.E.; Mahley, R.W. Differential cellular accumulation/retention of apolipoprotein E mediated by cell surface heparan sulfate proteoglycans. Apolipoproteins E3 and E2 greater than e4. J. Biol. Chem. 1998, 273, 13452–13460. [Google Scholar] [CrossRef]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 1999, 40, 1–16. [Google Scholar] [CrossRef]
- Michikawa, M.; Fan, Q.W.; Isobe, I.; Yanagisawa, K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 2000, 74, 1008–1016. [Google Scholar] [CrossRef]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Maiorano, J.N.; Dhanasekaran, P.; Ryan, R.O.; Phillips, M.C.; Lund-Katz, S.; Davidson, W.S. Helix orientation of the functional domains in apolipoprotein e in discoidal high density lipoprotein particles. J. Biol. Chem. 2004, 279, 14273–14279. [Google Scholar] [CrossRef]
- Peters-Libeu, C.A.; Newhouse, Y.; Hatters, D.M.; Weisgraber, K.H. Model of biologically active apolipoprotein E bound to dipalmitoylphosphatidylcholine. J. Biol. Chem. 2006, 281, 1073–1079. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Zaiou, M.; Wehrli, S.; Dhanasekaran, P.; Baldwin, F.; Weisgraber, K.H.; Phillips, M.C. Effects of lipid interaction on the lysine microenvironments in apolipoprotein E. J. Biol. Chem. 2000, 275, 34459–34464. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Zhong, N.; Rutenber, E.; Weisgraber, K.H. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J. Mol. Biol. 2006, 361, 932–944. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Beffert, U.; Aumont, N.; Dea, D.; Lussier-Cacan, S.; Davignon, J.; Poirier, J. Beta-amyloid peptides increase the binding and internalization of apolipoprotein E to hippocampal neurons. J. Neurochem. 1998, 70, 1458–1466. [Google Scholar] [CrossRef]
- Yamauchi, K.; Tozuka, M.; Hidaka, H.; Nakabayashi, T.; Sugano, M.; Katsuyama, T. Isoform-specific effect of apolipoprotein E on endocytosis of beta-amyloid in cultures of neuroblastoma cells. Ann. Clin. Lab. Sci. 2002, 32, 65–74. [Google Scholar]
- Nielsen, H.M.; Veerhuis, R.; Holmqvist, B.; Janciauskiene, S. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia 2009, 57, 978–988. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Ito, S.; Ohtsuki, S.; Kamiie, J.; Nezu, Y.; Terasaki, T. Cerebral clearance of human amyloid-beta peptide (1-40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 2007, 103, 2482–2490. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Basak, J.M.; Verghese, P.B.; Yoon, H.; Kim, J.; Holtzman, D.M. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J. Biol. Chem. 2012, 287, 13959–13971. [Google Scholar] [CrossRef]
- Tokuda, T.; Calero, M.; Matsubara, E.; Vidal, R.; Kumar, A.; Permanne, B.; Zlokovic, B.; Smith, J.D.; Ladu, M.J.; Rostagno, A.; et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem. J. 2000, 348 Pt 2, 359–365. [Google Scholar] [CrossRef]
- Sanan, D.A.; Weisgraber, K.H.; Russell, S.J.; Mahley, R.W.; Huang, D.; Saunders, A.; Schmechel, D.; Wisniewski, T.; Frangione, B.; Roses, A.D.; et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Investig. 1994, 94, 860–869. [Google Scholar] [CrossRef]
- Pillot, T.; Goethals, M.; Najib, J.; Labeur, C.; Lins, L.; Chambaz, J.; Brasseur, R.; Vandekerckhove, J.; Rosseneu, M. Beta-amyloid peptide interacts specifically with the carboxy-terminal domain of human apolipoprotein E: Relevance to Alzheimer’s disease. J. Neurochem. 1999, 72, 230–237. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, H.; Horie, T.; Burchett, J.M.; Restivo, J.L.; Rotllan, N.; Ramirez, C.M.; Verghese, P.B.; Ihara, M.; Hoe, H.S.; et al. microRNA-33 Regulates ApoE Lipidation and Amyloid-beta Metabolism in the Brain. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 14717–14726. [Google Scholar] [CrossRef]
- Boehm-Cagan, A.; Bar, R.; Liraz, O.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J. Alzheimer’s Dis. JAD 2016, 54, 1219–1233. [Google Scholar] [CrossRef]
- Rawat, V.; Wang, S.; Sima, J.; Bar, R.; Liraz, O.; Gundimeda, U.; Parekh, T.; Chan, J.; Johansson, J.O.; Tang, C.; et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 9611–9622. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef]
- Duan, Y.; Ye, T.; Qu, Z.; Chen, Y.; Miranda, A.; Zhou, X.; Lok, K.C.; Chen, Y.; Fu, A.K.Y.; Gradinaru, V.; et al. Brain-wide Cas9-mediated cleavage of a gene causing familial Alzheimer’s disease alleviates amyloid-related pathologies in mice. Nat. Biomed. Eng. 2022, 6, 168–180. [Google Scholar] [CrossRef]
- Liu, R.Y.; Gu, R.; Qi, X.L.; Zhang, T.; Zhao, Y.; He, Y.; Pei, J.J.; Guan, Z.Z. Decreased nicotinic receptors and cognitive deficit in rats intracerebroventricularly injected with beta-amyloid peptide(1-42) and fed a high-cholesterol diet. J. Neurosci. Res. 2008, 86, 183–193. [Google Scholar] [CrossRef]
- Xiu, J.; Nordberg, A.; Qi, X.; Guan, Z.Z. Influence of cholesterol and lovastatin on alpha-form of secreted amyloid precursor protein and expression of alpha7 nicotinic receptor on astrocytes. Neurochem. Int. 2006, 49, 459–465. [Google Scholar] [CrossRef]
- Ahmed, T.; Zahid, S.; Mahboob, A.; Farhat, S.M. Cholinergic System and Post-translational Modifications: An Insight on the Role in Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 480–494. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef]
- Karami, A.; Eriksdotter, M.; Kadir, A.; Almkvist, O.; Nordberg, A.; Darreh-Shori, T. CSF Cholinergic Index, a New Biomeasure of Treatment Effect in Patients With Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 12, 239. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L., 3rd; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Banuelos, C.; LaSarge, C.L.; McQuail, J.A.; Hartman, J.J.; Gilbert, R.J.; Ormerod, B.K.; Bizon, J.L. Age-related changes in rostral basal forebrain cholinergic and GABAergic projection neurons: Relationship with spatial impairment. Neurobiol. Aging 2013, 34, 845–862. [Google Scholar] [CrossRef]
- Kalmbach, A.; Hedrick, T.; Waters, J. Selective optogenetic stimulation of cholinergic axons in neocortex. J. Neurophysiol. 2012, 107, 2008–2019. [Google Scholar] [CrossRef]
- Zhu, H.; Yan, H.; Tang, N.; Li, X.; Pang, P.; Li, H.; Chen, W.; Guo, Y.; Shu, S.; Cai, Y.; et al. Impairments of spatial memory in an Alzheimer’s disease model via degeneration of hippocampal cholinergic synapses. Nat. Commun. 2017, 8, 1676. [Google Scholar] [CrossRef]
- Marsh, J.; Alifragis, P. Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616–623. [Google Scholar]
- Yan, H.; Pang, P.; Chen, W.; Zhu, H.; Henok, K.A.; Li, H.; Wu, Z.; Ke, X.; Wu, J.; Zhang, T.; et al. The Lesion Analysis of Cholinergic Neurons in 5XFAD Mouse Model in the Three-Dimensional Level of Whole Brain. Mol. Neurobiol. 2018, 55, 4115–4125. [Google Scholar]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Laursen, B.; Mork, A.; Plath, N.; Kristiansen, U.; Bastlund, J.F. Cholinergic degeneration is associated with increased plaque deposition and cognitive impairment in APPswe/PS1dE9 mice. Behav. Brain Res. 2013, 240, 146–152. [Google Scholar] [CrossRef]
- Vaucher, E.; Hamel, E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: Electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J. Neurosci. 1995, 15, 7427–7441. [Google Scholar] [CrossRef]
- Lecrux, C.; Sandoe, C.H.; Neupane, S.; Kropf, P.; Toussay, X.; Tong, X.K.; Lacalle-Aurioles, M.; Shmuel, A.; Hamel, E. Impact of Altered Cholinergic Tones on the Neurovascular Coupling Response to Whisker Stimulation. J. Neurosci. 2017, 37, 1518–1531. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, C.E.; Clark, L.R.; Dev, S.I.; Shin, D.D.; Jurick, S.M.; Rissman, R.A.; Liu, T.T.; Bondi, M.W. Interaction of age and APOE genotype on cerebral blood flow at rest. J. Alzheimer’s Dis. 2013, 34, 921–935. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bernstein, M.A.; Fox, N.C.; Thompson, P.; Alexander, G.; Harvey, D.; Borowski, B.; Britson, P.J.; Whitwell, J.L.; Ward, C.; et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J. Magn. Reson. Imaging 2008, 27, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Perez, J.M.; Evans, A.C.; Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Steinecke, A.; Bolton, M.M.; Taniguchi, H. Neuromodulatory control of inhibitory network arborization in the developing postnatal neocortex. Sci. Adv. 2022, 8, eabe7192. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zou, L.; Meng, L.; Qiang, G.; Yan, M.; Zhang, Z. Cholesterol Metabolism in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2021, 58, 2183–2201. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Gotz, J.; Schild, A.; Hoerndli, F.; Pennanen, L. Amyloid-induced neurofibrillary tangle formation in Alzheimer’s disease: Insight from transgenic mouse and tissue-culture models. Int. J. Dev. Neurosci. 2004, 22, 453–465. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Small, D.H.; Michaelson, S.; Sberna, G. Non-classical actions of cholinesterases: Role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 1996, 28, 453–483. [Google Scholar] [CrossRef]
- Baenziger, J.E.; Morris, M.L.; Darsaut, T.E.; Ryan, S.E. Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 2000, 275, 777–784. [Google Scholar] [CrossRef]
- Barrantes, F.J. Cholesterol effects on nicotinic acetylcholine receptor: Cellular aspects. Subcell. Biochem. 2010, 51, 467–487. [Google Scholar]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef]
- Bruses, J.L.; Chauvet, N.; Rutishauser, U. Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J. Neurosci. 2001, 21, 504–512. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta 2014, 1838, 532–545. [Google Scholar] [CrossRef]
- Pena, V.B.; Bonini, I.C.; Antollini, S.S.; Kobayashi, T.; Barrantes, F.J. alpha 7-type acetylcholine receptor localization and its modulation by nicotine and cholesterol in vascular endothelial cells. J. Cell Biochem. 2011, 112, 3276–3288. [Google Scholar] [CrossRef]
- Borroni, V.; Barrantes, F.J. Cholesterol modulates the rate and mechanism of acetylcholine receptor internalization. J. Biol. Chem. 2011, 286, 17122–17132. [Google Scholar] [CrossRef]
- Kumari, S.; Borroni, V.; Chaudhry, A.; Chanda, B.; Massol, R.; Mayor, S.; Barrantes, F.J. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J. Cell Biol. 2008, 181, 1179–1193. [Google Scholar] [CrossRef]
- Colon-Saez, J.O.; Yakel, J.L. The alpha7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 2011, 589 Pt 13, 3163–3174. [Google Scholar] [CrossRef]
- Hernandez, C.M.; Kayed, R.; Zheng, H.; Sweatt, J.D.; Dineley, K.T. Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2010, 30, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Juszczyk, G.; Mikulska, J.; Kasperek, K.; Pietrzak, D.; Mrozek, W.; Herbet, M. Chronic Stress and Oxidative Stress as Common Factors of the Pathogenesis of Depression and Alzheimer’s Disease: The Role of Antioxidants in Prevention and Treatment. Antioxidants 2021, 10, 1439. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D.; Mahady, L.; Perez, S.E.; Massa, S.M.; Longo, F.M.; Ikonomovic, M.D. Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 533. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of The Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029113. [Google Scholar] [CrossRef] [PubMed]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Se Thoe, E.; Fauzi, A.; Tang, Y.Q.; Chamyuang, S.; Chia, A.Y.Y. A review on advances of treatment modalities for Alzheimer’s disease. Life Sci. 2021, 276, 119129. [Google Scholar] [CrossRef]
- Samant, N.P.; Gupta, G.L. Novel therapeutic strategies for Alzheimer’s disease targeting brain cholesterol homeostasis. Eur. J. Neurosci. 2021, 53, 673–686. [Google Scholar] [CrossRef]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef]
- Zhao, L.; Xiao, Y.; Xiu, J.; Tan, L.C.; Guan, Z.Z. Protection against the Neurotoxic Effects of beta-Amyloid Peptide on Cultured Neuronal Cells by Lovastatin Involves Elevated Expression of alpha7 Nicotinic Acetylcholine Receptors and Activating Phosphorylation of Protein Kinases. Am. J. Pathol. 2018, 188, 1081–1093. [Google Scholar] [CrossRef]
- Borroni, V.; Kamerbeek, C.; Pediconi, M.F.; Barrantes, F.J. Lovastatin Differentially Regulates alpha7 and alpha4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons. Molecules 2020, 25, 4838. [Google Scholar] [CrossRef]
- Dehnavi, S.; Kiani, A.; Sadeghi, M.; Biregani, A.F.; Banach, M.; Atkin, S.L.; Jamialahmadi, T.; Sahebkar, A. Targeting AMPK by Statins: A Potential Therapeutic Approach. Drugs 2021, 81, 923–933. [Google Scholar] [CrossRef]
- Li, H.H.; Lin, C.L.; Huang, C.N. Neuroprotective effects of statins against amyloid beta-induced neurotoxicity. Neural Regen. Res. 2018, 13, 198–206. [Google Scholar]
- Zhao, L.; Chen, T.; Wang, C.; Li, G.; Zhi, W.; Yin, J.; Wan, Q.; Chen, L. Atorvastatin in improvement of cognitive impairments caused by amyloid beta in mice: Involvement of inflammatory reaction. BMC Neurol. 2016, 16, 18. [Google Scholar] [CrossRef]
- Wang, C.; Chen, T.; Li, G.; Zhou, L.; Sha, S.; Chen, L. Simvastatin prevents beta-amyloid(25-35)-impaired neurogenesis in hippocampal dentate gyrus through alpha7nAChR-dependent cascading PI3K-Akt and increasing BDNF via reduction of farnesyl pyrophosphate. Neuropharmacology 2015, 97, 122–132. [Google Scholar] [CrossRef]
- Gao, K.; Shen, Z.; Yuan, Y.; Han, D.; Song, C.; Guo, Y.; Mei, X. Simvastatin inhibits neural cell apoptosis and promotes locomotor recovery via activation of Wnt/beta-catenin signaling pathway after spinal cord injury. J. Neurochem. 2016, 138, 139–149. [Google Scholar] [CrossRef]
- Tong, X.K.; Royea, J.; Hamel, E. Simvastatin rescues memory and granule cell maturation through the Wnt/beta-catenin signaling pathway in a mouse model of Alzheimer’s disease. Cell Death Dis. 2022, 13, 325. [Google Scholar] [CrossRef]
- Kurinami, H.; Sato, N.; Shinohara, M.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Ogihara, T.; Morishita, R. Prevention of amyloid beta-induced memory impairment by fluvastatin, associated with the decrease in amyloid beta accumulation and oxidative stress in amyloid beta injection mouse model. Int. J. Mol. Med. 2008, 21, 531–537. [Google Scholar]
- Sodero, A.O.; Barrantes, F.J. Pleiotropic effects of statins on brain cells. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183340. [Google Scholar] [CrossRef]
- Kornelius, E.; Li, H.H.; Peng, C.H.; Hsiao, H.W.; Yang, Y.S.; Huang, C.N.; Lin, C.L. Mevastatin promotes neuronal survival against Abeta-induced neurotoxicity through AMPK activation. Metab. Brain Dis. 2017, 32, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.; Chuang, Y.S.; Hsieh, H.M.; Lee, T.C.; Chiu, K.F.; Liu, C.K.; Wu, M.T. Early Statin Use and the Progression of Alzheimer Disease: A Total Population-Based Case-Control Study. Medicine 2015, 94, e2143. [Google Scholar] [CrossRef] [PubMed]
- Geifman, N.; Brinton, R.D.; Kennedy, R.E.; Schneider, L.S.; Butte, A.J. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Davey, A.K.; McDermott, C.M.; Grant, G.D.; Lewohl, J.; Anoopkumar-Dukie, S. Differences in statin associated neuroprotection corresponds with either decreased production of IL-1beta or TNF-alpha in an in vitro model of neuroinflammation-induced neurodegeneration. Toxicol. Appl. Pharmacol. 2018, 344, 56–73. [Google Scholar] [CrossRef]
- Imamura, O.; Arai, M.; Dateki, M.; Oishi, K.; Takishima, K. Donepezil-induced oligodendrocyte differentiation is mediated through estrogen receptors. J. Neurochem. 2020, 155, 494–507. [Google Scholar] [CrossRef]
- Moreira, N.; Lima, J.; Marchiori, M.F.; Carvalho, I.; Sakamoto-Hojo, E.T. Neuroprotective Effects of Cholinesterase Inhibitors: Current Scenario in Therapies for Alzheimer’s Disease and Future Perspectives. J. Alzheimer’s Dis. Rep. 2022, 6, 177–193. [Google Scholar] [CrossRef]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients With Alzheimer’s Disease. JAMA 1994, 271, 7. [Google Scholar] [CrossRef]
- Khoobi, M.; Ghanoni, F.; Nadri, H.; Moradi, A.; Pirali Hamedani, M.; Homayouni Moghadam, F.; Emami, S.; Vosooghi, M.; Zadmard, R.; Foroumadi, A.; et al. New tetracyclic tacrine analogs containing pyrano [2,3-c]pyrazole: Efficient synthesis, biological assessment and docking simulation study. Eur. J. Med. Chem. 2015, 89, 296–303. [Google Scholar] [CrossRef]
- Chioua, M.; Perez-Pena, J.; Garcia-Font, N.; Moraleda, I.; Iriepa, I.; Soriano, E.; Marco-Contelles, J.; Oset-Gasque, M.J. Pyranopyrazolotacrines as nonneurotoxic, Abeta-anti-aggregating and neuroprotective agents for Alzheimer’s disease. Future Med. Chem. 2015, 7, 845–855. [Google Scholar] [CrossRef]
- Zheng, H.; Niu, S.; Zhao, H.; Li, S.; Jiao, J. Donepezil improves the cognitive impairment in a tree shrew model of Alzheimer’s disease induced by amyloid-beta1-40 via activating the BDNF/TrkB signal pathway. Metab. Brain Dis. 2018, 33, 1961–1974. [Google Scholar] [CrossRef]
- Dubois, B.; Chupin, M.; Hampel, H.; Lista, S.; Cavedo, E.; Croisile, B.; Louis Tisserand, G.; Touchon, J.; Bonafe, A.; Ousset, P.J.; et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1041–1049. [Google Scholar] [CrossRef]
- Cavedo, E.; Dubois, B.; Colliot, O.; Lista, S.; Croisile, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; Rouaud, O.; et al. Reduced Regional Cortical Thickness Rate of Change in Donepezil-Treated Subjects With Suspected Prodromal Alzheimer’s Disease. J. Clin. Psychiatry 2016, 77, e1631–e1638. [Google Scholar] [CrossRef]
- Schmitz, T.W.; Nathan Spreng, R.; Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef]
- Cavedo, E.; Grothe, M.J.; Colliot, O.; Lista, S.; Chupin, M.; Dormont, D.; Houot, M.; Lehericy, S.; Teipel, S.; Dubois, B.; et al. Reduced basal forebrain atrophy progression in a randomized Donepezil trial in prodromal Alzheimer’s disease. Sci. Rep. 2017, 7, 11706. [Google Scholar] [CrossRef]
- Terada, K.; Migita, K.; Matsushima, Y.; Sugimoto, Y.; Kamei, C.; Matsumoto, T.; Mori, M.; Matsunaga, K.; Takata, J.; Karube, Y. Cholinesterase inhibitor rivastigmine enhances nerve growth factor-induced neurite outgrowth in PC12 cells via sigma-1 and sigma-2 receptors. PLoS ONE 2018, 13, e0209250. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef]
- Potkin, S.G.; Anand, R.; Fleming, K.; Alva, G.; Keator, D.; Carreon, D.; Messina, J.; Wu, J.C.; Hartman, R.; Fallon, J.H. Brain metabolic and clinical effects of rivastigmine in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2001, 4, 223–230. [Google Scholar] [CrossRef]
- Ferris, S.; Nordberg, A.; Soininen, H.; Darreh-Shori, T.; Lane, R. Progression from mild cognitive impairment to Alzheimer’s disease: Effects of sex, butyrylcholinesterase genotype, and rivastigmine treatment. Pharm. Genom. 2009, 19, 635–646. [Google Scholar] [CrossRef]
- Moss, D.E.; Perez, R.G. Anti-Neurodegenerative Benefits of Acetylcholinesterase Inhibitors in Alzheimer’s Disease: Nexus of Cholinergic and Nerve Growth Factor Dysfunction. Curr. Alzheimer Res. 2021, 18, 1010–1022. [Google Scholar] [CrossRef]
- Moss, D.E. Is Combining an Anticholinergic with a Cholinesterase Inhibitor a Good Strategy for High-Level CNS Cholinesterase Inhibition? J. Alzheimer’s Dis. 2019, 71, 1099–1103. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Baakman, A.C.; Gavan, C.; van Doeselaar, L.; de Kam, M.; Broekhuizen, K.; Bajenaru, O.; Camps, L.; Swart, E.L.; Kalisvaart, K.; Schoonenboom, N.; et al. Acute response to cholinergic challenge predicts long-term response to galantamine treatment in patients with Alzheimer’s disease. Br. J. Clin. Pharmacol. 2022, 88, 2814–2829. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Pentz, R.; Hall, H. The Brain NGF Metabolic Pathway in Health and in Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Gong, H.; Xiao, H.; Petersen, R.B.; Zheng, L.; Huang, K. Inhibiting toxic aggregation of amyloidogenic proteins: A therapeutic strategy for protein misfolding diseases. Biochim. Biophys. Acta 2013, 1830, 4860–4871. [Google Scholar] [CrossRef] [PubMed]
- Goasdoue, K.; Miller, S.M.; Colditz, P.B.; Bjorkman, S.T. Review: The blood-brain barrier; protecting the developing fetal brain. Placenta 2017, 54, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.H.; Mir, M.; Ngowi, E.E.; Zafar, U.; Khakwani, M.; Khattak, S.; Zhai, Y.K.; Jiang, E.S.; Zheng, M.; Duan, S.F.; et al. Nanomedicine: A Promising Way to Manage Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2021, 9, 630055. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.T.; Ma, C.; Li, G.J.; Zheng, X.Y.; Hao, Y.T.; Yang, Y.; Wang, X. Application of Antibody Fragments Against Abeta With Emphasis on Combined Application With Nanoparticles in Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 654611. [Google Scholar] [CrossRef]
- Zhang, F.Q.; Jiang, J.L.; Zhang, J.T.; Niu, H.; Fu, X.Q.; Zeng, L.L. Current status and future prospects of stem cell therapy in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 242–250. [Google Scholar]
- Cristina Buzea, I.P. Nanomaterials and Their Classification; Springer: Delhi, India, 2017. [Google Scholar]
- Zhang, Y.; Zhu, J. Synthesis and characterization of several one-dimensional nanomaterials. Micron 2002, 33, 523–534. [Google Scholar] [CrossRef]
- Pentlavalli, S.; Coulter, S.; Laverty, G. Peptide Nanomaterials for Drug Delivery Applications. Curr. Protein Pept. Sci. 2020, 21, 401–412. [Google Scholar] [CrossRef]
- Beaux, M.F., 2nd; McIlroy, D.N.; Gustin, K.E. Utilization of solid nanomaterials for drug delivery. Expert Opin. Drug Deliv. 2008, 5, 725–735. [Google Scholar] [CrossRef]
- Zorkina, Y.; Abramova, O.; Ushakova, V.; Morozova, A.; Zubkov, E.; Valikhov, M.; Melnikov, P.; Majouga, A.; Chekhonin, V. Nano Carrier Drug Delivery Systems for the Treatment of Neuropsychiatric Disorders: Advantages and Limitations. Molecules 2020, 25, 5294. [Google Scholar] [CrossRef]
- Binda, A.; Murano, C.; Rivolta, I. Innovative Therapies and Nanomedicine Applications for the Treatment of Alzheimer’s Disease: A State-of-the-Art (2017–2020). Int. J. Nanomed. 2020, 15, 6113–6135. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Sajadimajd, S.; Jafari, S.; Izadi, Z.; Sarvari, S.; Sharifi, M.; Falahati, M.; Moakedi, F.; Muganda, W.C.A.; Muller, M.; et al. Novel therapeutic strategies for Alzheimer’s disease: Implications from cell-based therapy and nanotherapy. Nanomedicine 2020, 24, 102149. [Google Scholar] [CrossRef]
- Silva, S.; Marto, J.; Goncalves, L.; Almeida, A.J.; Vale, N. Formulation, Characterization and Evaluation against SH-SY5Y Cells of New Tacrine and Tacrine-MAP Loaded with Lipid Nanoparticles. Nanomaterials 2020, 10, 2089. [Google Scholar] [CrossRef]
- Shamarekh, K.S.; Gad, H.A.; Soliman, M.E.; Sammour, O.A. Development and evaluation of protamine-coated PLGA nanoparticles for nose-to-brain delivery of tacrine: In-vitro and in-vivo assessment. J. Drug Deliv. Sci. Technol. 2020, 13, 101724. [Google Scholar] [CrossRef]
- Joe, V.F.a.S.S.K. Formulation, characterization and determination of anti-alzheimeric activity of tacrine loaded poly (lactide-co-glycolide) nanoparticles. J. Pharm. Sci. Res. 2018, 9, 10. [Google Scholar]
- AnjiReddy, K.; Karpagam, S. In Vitro and In Vivo Evaluation of Oral Disintegrating Nanofiber and Thin-Film Contains Hyperbranched Chitosan/Donepezil for Active Drug Delivery. J. Polym. Environ. 2021, 29, 922–936. [Google Scholar] [CrossRef]
- Topal, G.R.; Meszaros, M.; Porkolab, G.; Szecsko, A.; Polgar, T.F.; Siklos, L.; Deli, M.A.; Veszelka, S.; Bozkir, A. ApoE-Targeting Increases the Transfer of Solid Lipid Nanoparticles with Donepezil Cargo across a Culture Model of the Blood-Brain Barrier. Pharmaceutics 2020, 13, 38. [Google Scholar] [CrossRef]
- Mendes, I.T.; Ruela, A.L.M.; Carvalho, F.C.; Freitas, J.T.J.; Bonfilio, R.; Pereira, G.R. Development and characterization of nanostructured lipid carrier-based gels for the transdermal delivery of donepezil. Colloids Surf. B Biointerfaces 2019, 177, 274–281. [Google Scholar] [CrossRef]
- El-Assal, M.I.; Samuel, D. Optimization of rivastigmine chitosan nanoparticles for neurodegenerative alzheimer. In vitro and ex vivo characterizations. Int. J. Pharm. Pharm. Sci. 2022, 14, 11. [Google Scholar] [CrossRef]
- Srinivas Hebbar, M.P. Amitha Shetty, Akhilesh Dubey, In vitro Investigation of Conventional, Chitosan Coated and Electrosteric Stealth Liposomes of Rivastigmine Tartrate for the treatment of Alzheimer’s Disease. Int. J. Pharm. Investig. 2020, 10, 6. [Google Scholar]
- Bhanderi, M.; Shah, J.; Gorain, B.; Nair, A.B.; Jacob, S.; Asdaq, S.M.B.; Fattepur, S.; Alamri, A.S.; Alsanie, W.F.; Alhomrani, M.; et al. Optimized Rivastigmine Nanoparticles Coated with Eudragit for Intranasal Application to Brain Delivery: Evaluation and Nasal Ciliotoxicity Studies. Materials 2021, 14, 6291. [Google Scholar] [CrossRef]
- Kandil, L.S.; Farid, R.M.; ElGamal, S.S.; Hanafy, A.S. Intranasal galantamine/chitosan complex nanoparticles elicit neuroprotection potentials in rat brains via antioxidant effect. Drug Dev. Ind. Pharm. 2021, 47, 735–740. [Google Scholar] [CrossRef]
- Georgieva, D.; Ivanova-Mileva, K.; Ivanova, S.; Kostova, B.; Rachev, D.; Christova, D. Thermoresponsive poly(N-isopropylacrylamide) copolymer networks for galantamine hydrobromide delivery. Colloid Polym. Sci. 2020, 8, 377–384. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.-M. Modulating the aggregation of amyloid proteins by macrocycles. Aggregaate 2022, 3, e161. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X.; Pan, Y.C.; Yan, Y.; Hu, X.Y.; Chen, R.; Ravoo, B.J.; Guo, D.S.; Zhang, T. Recognition and Removal of Amyloid-beta by a Heteromultivalent Macrocyclic Coassembly: A Potential Strategy for the Treatment of Alzheimer’s Disease. Adv. Mater. 2021, 33, e2006483. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jia, S.; Wang, W.; Yuan, Z.; Jan Ravoo, B.; Guo, D.S. Heteromultivalent peptide recognition by co-assembly of cyclodextrin and calixarene amphiphiles enables inhibition of amyloid fibrillation. Nat. Chem. 2019, 11, 86–93. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statin | Evidence | Model | Reference |
|---|---|---|---|
| Lovastatin | Decreases neurotoxic effect of Aβ, favors non-amyloidogenic processing, upregulates the expression of α7 nAChR and the stimulation of extracellular signal-regulated kinase phosphorylation (ERK), activates Akt signaling pathway, which in turn, inhibits downstream GSK-3β and decreases tau hyperphosphorylation and aggregation. | SH-SY5Y cells. Rat hippocampal neuronal cells | [212,214,218] |
| Atorvastatin | Improves inflammatory impairment, depresses inflammatory responses, and prevents Aβ25–35-induced neurotoxicity. | Mice injected with Aβ 25–35 intracerebroventricularly. Cultured hippocampal neurons | [217] |
| Simvastatin | Improves spatial cognitive function, protects neurogenesis through α7nAChR-cascading PI3K-Akt, increases BDNF levels, reduces neuronal apoptosis, improves functional and pathological recovery by activating the Wnt/β-catenin signaling pathway. | Mice injected with Aβ 25-35 intracerebroventricularly. Spinal cord injury rat model. APP mice model. | [213,215,216] |
| Fluvastatin | Decreases Aβ accumulation, and prevents cognitive deterioration, oxidative stress and loss of neurons in the basal forebrain induced by Aβ25-35. | Mice injected with Aβ 25-35 intracerebroventricularly. | [220,221] |
| Mevastatin | Restores insulin signaling and protects amyloid-induced neurotoxicity through activation of AMP-activated protein kinase (AMPK). | SK-N-MC neuronal cells. | [219] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Peña, V.; Pichardo-Rojas, P.; Sánchez-Barbosa, T.; Ortíz-Islas, E.; Rodríguez-Pérez, C.E.; Montes, P.; Ramos-Palacios, G.; Silva-Adaya, D.; Valencia-Quintana, R.; Cerna-Cortes, J.F.; et al. Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12092. https://doi.org/10.3390/ijms232012092
Campos-Peña V, Pichardo-Rojas P, Sánchez-Barbosa T, Ortíz-Islas E, Rodríguez-Pérez CE, Montes P, Ramos-Palacios G, Silva-Adaya D, Valencia-Quintana R, Cerna-Cortes JF, et al. Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(20):12092. https://doi.org/10.3390/ijms232012092
Chicago/Turabian StyleCampos-Peña, Victoria, Pavel Pichardo-Rojas, Talía Sánchez-Barbosa, Emma Ortíz-Islas, Citlali Ekaterina Rodríguez-Pérez, Pedro Montes, Gerardo Ramos-Palacios, Daniela Silva-Adaya, Rafael Valencia-Quintana, Jorge Francisco Cerna-Cortes, and et al. 2022. "Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 20: 12092. https://doi.org/10.3390/ijms232012092
APA StyleCampos-Peña, V., Pichardo-Rojas, P., Sánchez-Barbosa, T., Ortíz-Islas, E., Rodríguez-Pérez, C. E., Montes, P., Ramos-Palacios, G., Silva-Adaya, D., Valencia-Quintana, R., Cerna-Cortes, J. F., & Toral-Rios, D. (2022). Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease. International Journal of Molecular Sciences, 23(20), 12092. https://doi.org/10.3390/ijms232012092