
Intersection of the Ubiquitin–Proteasome System with Oxidative Stress in Cardiovascular Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. UPS and Oxidative Stress
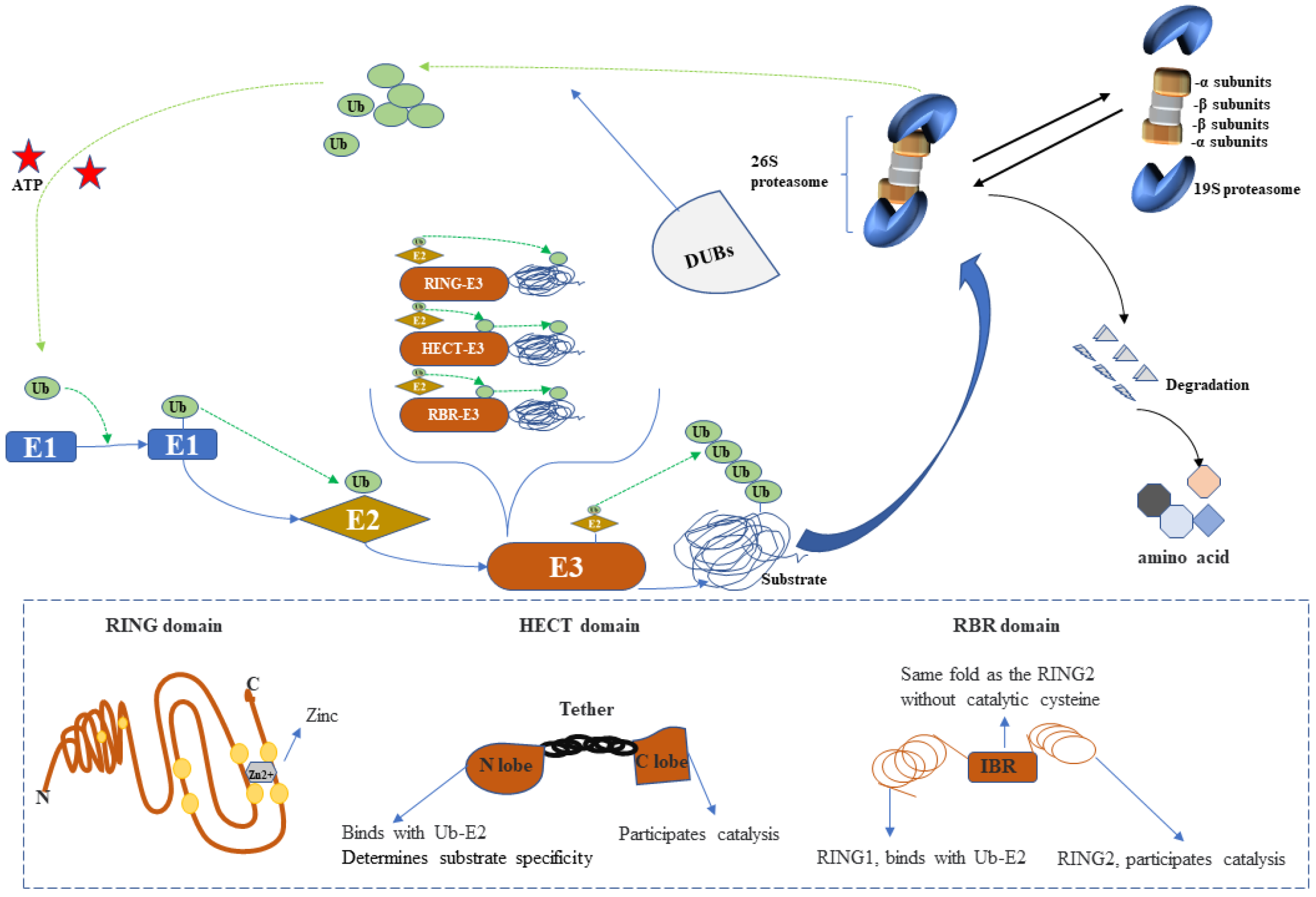
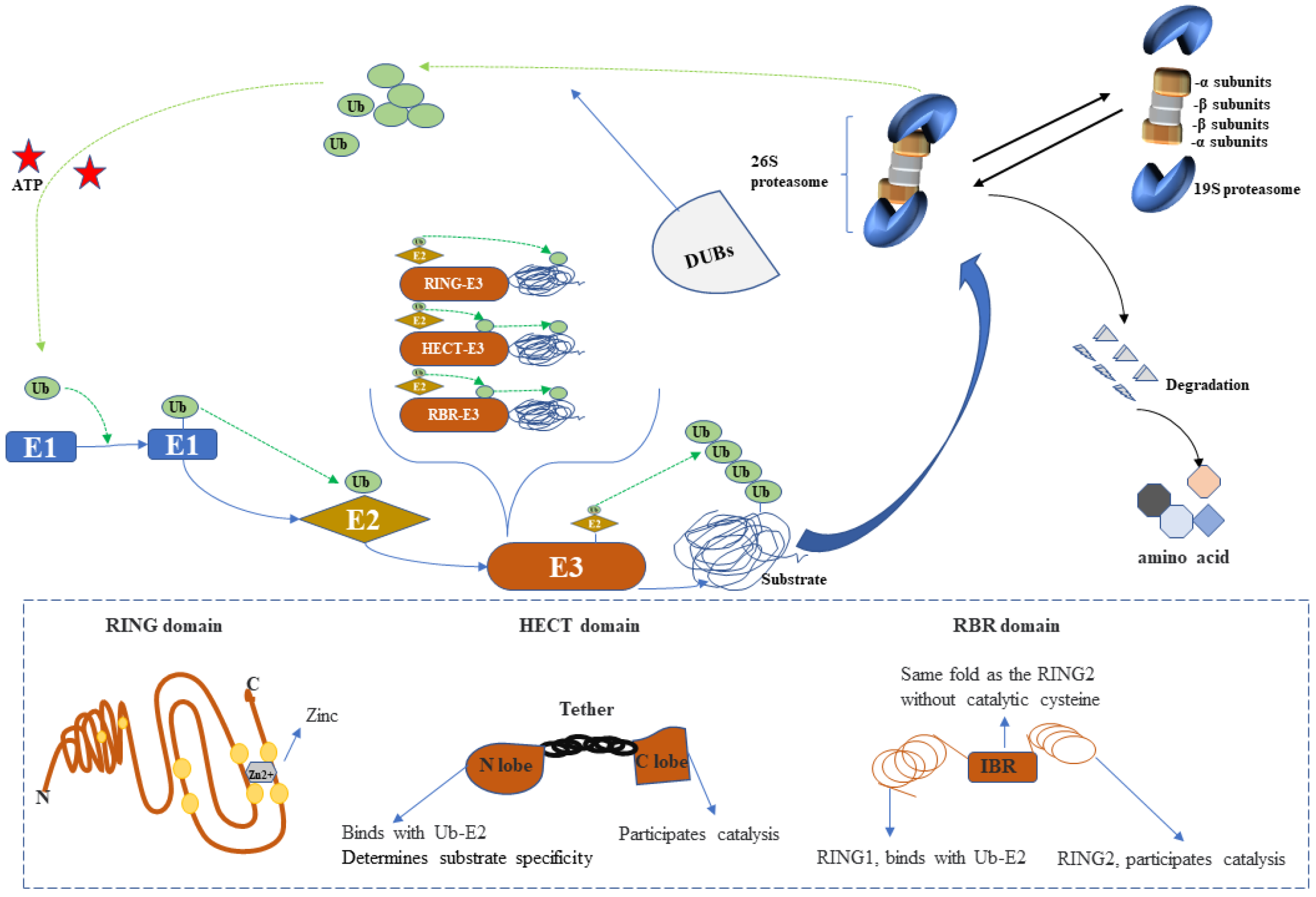
2.1. UPS
2.2. Oxidative Stress
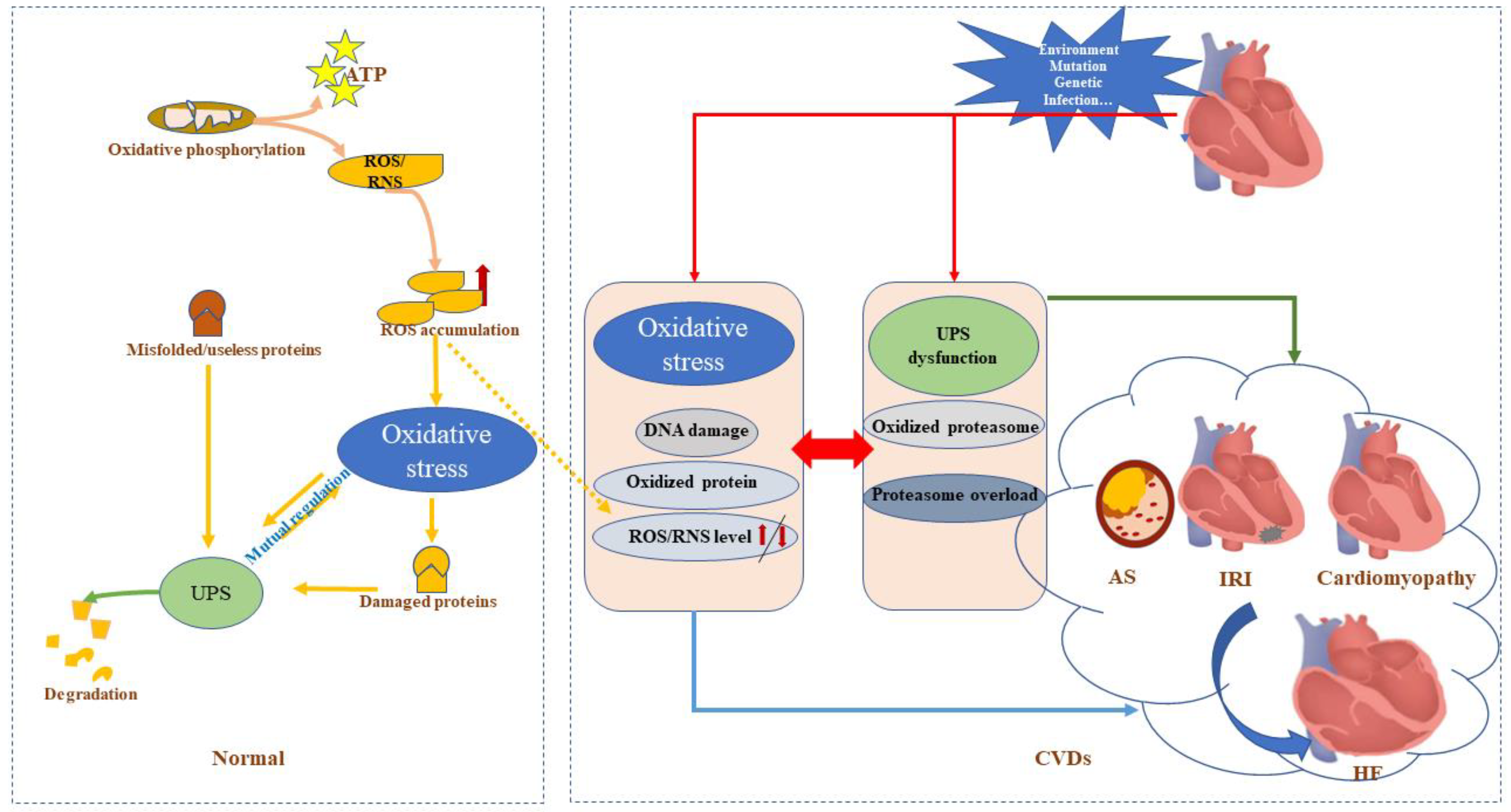
2.3. Oxidative Stress Directly Affects the UPS
2.4. Role of the UPS in Oxidative Stress
3. Interaction of the UPS and Oxidative Stress in CVDs
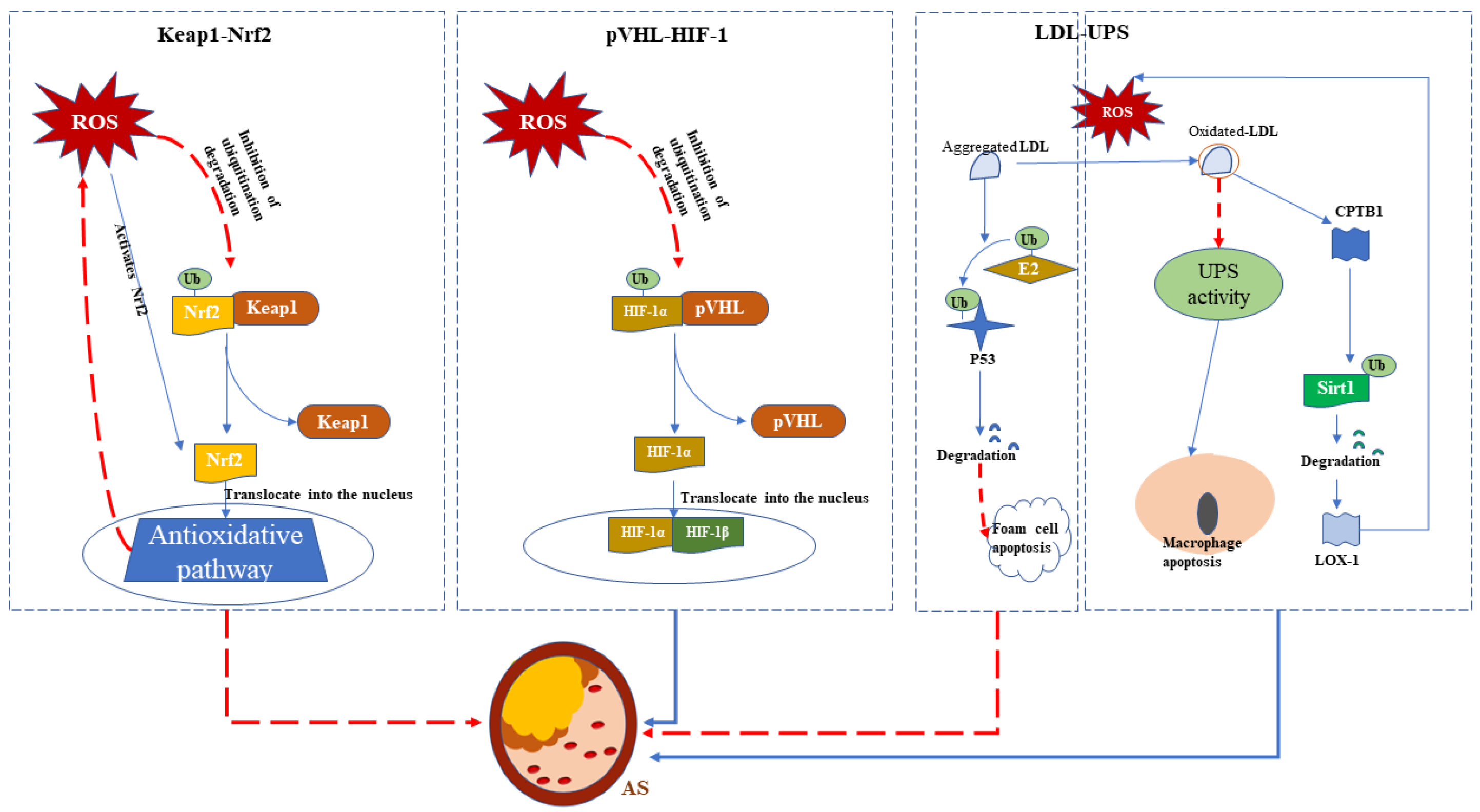
3.1. Atherosclerosis
3.2. Ischemia–Reperfusion Injury
3.3. Cardiomyopathy
3.4. Heart Failure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amini, M.; Zayeri, F.; Salehi, M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: Results from global burden of disease study 2017. BMC Public Health 2021, 21, 401. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, e148554. [Google Scholar] [CrossRef]
- Dzau, V.J.; Antman, E.M.; Black, H.R.; Hayes, D.L.; Manson, J.E.; Plutzky, J.; Popma, J.J.; Stevenson, W. The cardiovascular disease continuum validated: Clinical evidence of improved patient outcomes: Part I: Pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease). Circulation 2006, 114, 2850–2870. [Google Scholar] [CrossRef]
- Abdellatif, M.; Ljubojevic-Holzer, S.; Madeo, F.; Sedej, S. Autophagy in cardiovascular health and disease. Rog. Mol. Biol. Transl. Sci. 2020, 172, 87–106. [Google Scholar] [CrossRef]
- Organization, Cardiovascular Diseases (CVDs)—World Health Organization. Definition of Cardiovascular Diseases. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 11 June 2021).
- Hyun, S.H.; Bhilare, K.D.; In, G.; Park, C.K.; Kim, J.H. Effects of Panax ginseng and ginsenosides on oxidative stress and car-diovascular diseases: Pharmacological and therapeutic roles. J. Ginseng Res. 2022, 46, 33–38. [Google Scholar] [CrossRef]
- da Silva, D.V.T.; Baião, D.D.S.; Ferreira, V.F.; Paschoalin, V.M.F. Betanin as a multipath oxidative stress and inflammation modulator: A beetroot pigment with protective effects on cardiovascular disease pathogenesis. Crit. Rev. Food Sci. Nutr. 2022, 62, 539–554. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol.-Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.; Catalgol, B.; Grune, T. The proteasomal system. Mol. Aspects Med. 2009, 30, 191–296. [Google Scholar] [CrossRef]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta 2012, 1824, 3–13. [Google Scholar] [CrossRef]
- Willis, M.S.; Bevilacqua, A.; Pulinilkunnil, T.; Kienesberger, P.; Tannu, M.; Patterson, C. The role of ubiquitin ligases in cardiac disease. J. Mol. Cell. Cardiol. 2014, 71, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.I.; Parry, T.L.; Willis, M.S. Ubiquitin Ligases and Posttranslational Regulation of Energy in the Heart: The Hand that Feeds. Compr. Physiol. 2017, 7, 841–862. [Google Scholar] [CrossRef]
- Goto, J.; Otaki, Y.; Watanabe, T.; Watanabe, M. The Role of HECT-Type E3 Ligase in the Development of Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 6065. [Google Scholar] [CrossRef]
- Demasi, M.; Netto, L.E.; Silva, G.M.; Hand, A.; de Oliveira, C.L.; Bicev, R.N.; Gozzo, F.; Barros, M.H.; Leme, J.M.; Ohara, E. Redox regulation of the proteasome via S-glutathionylation. Redox Biol. 2013, 2, 44–51. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Trempe, J.-F. Reading the ubiquitin postal code. Curr. Opin. Struct. Biol. 2011, 21, 792–801. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [Green Version]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Olguín, H.C. The Gentle Side of the UPS: Ubiquitin-Proteasome System and the Regulation of the Myogenic Program. Front. Cell Dev. Biol. 2021, 9, 821839. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e1. [Google Scholar] [CrossRef]
- Donohue, T.M., Jr. The ubiquitin-proteasome system and its role in ethanol-induced disorders. Addict. Biol. 2002, 7, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W.; Cejka, Z.; Kania, M.; Seemuller, E. The proteasome: A macromolecular assembly designed to confine proteolysis to a nanocompartment. Biol. Chem. 1997, 378, 121–130. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by Mitochondrial Oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [Green Version]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Gomes, A.V.; Zong, C.; Ping, P. Protein Degradation by the 26S Proteasome System in the Normal and Stressed Myocardium. Antioxid. Redox Signal. 2006, 8, 1677–1691. [Google Scholar] [CrossRef]
- Ishii, T.; Sakurai, T.; Usami, H.; Uchida, K. Oxidative Modification of Proteasome: Identification of an Oxidation-Sensitive Subunit in 26 S Proteasome. Biochemistry 2005, 44, 13893–13901. [Google Scholar] [CrossRef]
- Yang, W.; Chen, L.; Ding, Y.; Zhuang, X.; Kang, U.J. Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ-1-deficient mice. Hum. Mol. Genet. 2007, 16, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, E.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S Proteasome Disassembly upon Mitochondrial Stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1–Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, T.; Fujii, J. Emerging connections between oxidative stress, defective proteolysis, and metabolic diseases. Free Radic. Res. 2020, 54, 931–946. [Google Scholar] [CrossRef]
- Heckle, M.R.; Flatt, D.M.; Sun, Y.; Mancarella, S.; Marion, T.N.; Gerling, I.C.; Weber, K.T. Atrophied cardiomyocytes and their potential for rescue and recovery of ventricular function. Heart Fail. Rev. 2016, 21, 191–198. [Google Scholar] [CrossRef]
- Versari, D.; Herrmann, J.; Gössl, M.; Mannheim, D.; Sattler, K.; Meyer, F.B.; Lerman, L.O.; Lerman, A. Dysregulation of the Ubiquitin-Proteasome System in Human Carotid Atherosclerosis. Arter. Thromb. Vasc. Biol. 2006, 26, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Wilck, N.; Ludwig, A. Targeting the Ubiquitin-Proteasome System in Atherosclerosis: Status Quo, Challenges, and Perspectives. Antioxid. Redox Signal. 2014, 21, 2344–2363. [Google Scholar] [CrossRef]
- Martin, T.G.; Kirk, J.A. Under construction: The dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J. Mol. Cell. Cardiol. 2020, 148, 89–102. [Google Scholar] [CrossRef]
- Reyskens, K.M.; Essop, M.F. HIV protease inhibitors and onset of cardiovascular diseases: A central role for oxidative stress and dysregulation of the ubiquitin–proteasome system. Biochim. Biophys. Acta 2014, 1842, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chung, A.C.K.; Fan, R.; Lee, H.M.; Xu, G.; Tomlinson, B.; Chan, J.C.N.; Kong, A.P.S. Sirt3 Deficiency Increased the Vulnerability of Pancreatic Beta Cells to Oxidative Stress-Induced Dysfunction. Antioxid. Redox Signal. 2017, 27, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Lerman, L.O.; Lerman, A. On to the road to degradation: Atherosclerosis and the proteasome. Cardiovasc. Res. 2010, 85, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Kashirskikh, D.A.; Sukhorukov, V.N.; Kalmykov, V.; Omelchenko, A.V.; Orekhov, A.N. Cholesterol Transport Dysfunction and Its Involvement in Atherogenesis. Int. J. Mol. Sci. 2022, 23, 1332. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits athero-sclerotic lesion formation in ldl-receptor knockout mice. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia Inducible Factor-α Binding and Ubiquitylation by the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [Green Version]
- Brand, K.; Page, S.; Rogler, G.; Bartsch, A.; Brandl, R.; Knuechel, R.; Page, M.; Kaltschmidt, C.; Baeuerle, P.A.; Neumeier, D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Investig. 1996, 97, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Canty, T.G., Jr.; Boyle, E.M., Jr.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation 1999, 100 (Suppl. S19), II361–II364. [Google Scholar]
- Cullen, S.J.; Ponnappan, S.; Ponnappan, U. Proteasome inhibition up-regulates inflammatory gene transcription induced by an atypical pathway of NF-kappaB activation. Biochem. Pharmacol. 2010, 79, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, Y.; Zhao, S.; Sun, Y. Septin4 as a novel binding partner of PARP1 contributes to oxidative stress induced human umbilical vein endothelial cells injure. Biochem. Biophys. Res. Commun. 2018, 496, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Y.; Wu, B.; You, S.; Sun, Y. Role of WW domain E3 ubiquitin protein ligase 2 in modulating ubiquitination and Degradation of Septin4 in oxidative stress endothelial injury. Redox Biol. 2020, 30, 101419. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Furukawa, Y.; Kubo, N.; Tokura, A.; Hayashi, N.; Nakamura, M.; Matsuda, M.; Sakurabayashi, I. Induction of ubiquitin-conjugating enzyme by aggregated low density lipoprotein in human macrophages and its implications for athero-sclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Vieira, O.; Escargueil-Blanc, I.; Jürgens, G.; Borner, C.; Almeida, L.; Salvayre, R.; Nègre-Salvayre, A. Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: Potential role in oxidized LDL-induced apoptosis. FASEB J. 2000, 14, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Xiwen, L.; Ren, G.; Yin, D.; Guo, S.; Zhao, Y. Depletion of CPEB1 protects against oxidized LDL-induced endothelial apoptosis and inflammation though SIRT1/LOX-1 signalling pathway. Life Sci. 2019, 239, 116874. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Oikonomou, E.K.; Antoniades, C. Immunometabolic Regulation of Vascular Redox State: The Role of Adipose Tissue. Antioxid. Redox Signal. 2018, 29, 313–336. [Google Scholar] [CrossRef] [Green Version]
- Akoumianakis, I.; Sanna, F.; Margaritis, M.; Badi, I.; Akawi, N.; Herdman, L.; Coutinho, P.; Fagan, H.; Antonopoulos, A.S.; Oikonomou, E.K.; et al. Adipose tissue-derived WNT5A regulates vascular redox signaling in obesity via USP17/RAC1-mediated activation of NADPH oxidases. Sci. Transl. Med. 2019, 11, eaav5055. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Gersh, B.J. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: A continual challenge. Eur. Heart J. 2017, 38, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Wu, W.; Sui, L.; Huang, Q.; Nan, Y.; Liu, J.; Ai, K. Reactive oxygen species-based nanomaterials for the treatment of myocardial ischemia reperfusion injuries. Bioact. Mater. 2022, 7, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Gelman, S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesi-ology 2001, 94, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Pereyra, L.H.; Toledo, A.H.; Walsh, J.; Lopez-Neblina, F. Molecular signaling pathways in ischemia/reperfusion. Exp. Clin. Transplant. 2004, 2, 174–177. [Google Scholar] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Niu, H.-T.; Wang, P.-L.; Lu, J.; Zhao, H.; Liu, S.-H.; Zheng, Q.S.; Li, C.-G. Cardioprotective Effect of Licochalcone D against Myocardial Ischemia/Reperfusion Injury in Langendorff-Perfused Rat Hearts. PLoS ONE 2015, 10, e0128375. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhi-bition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Reg-ulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Cheng, L.; Gao, X.; Chen, S.; Wu, P.; Wang, C.; Liu, Z. Covalent modification of Keap1 at Cys77 and Cys434 by pubescenoside a suppresses oxidative stress-induced NLRP3 inflammasome activation in myocardial ischemia-reperfusion injury. Theranostics 2021, 11, 861–877. [Google Scholar] [CrossRef]
- Liu, C.-C.; Prior, J.; Piwnica-Worms, D.; Bu, G. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5141. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Z.; Li, Y.; Ma, L.; Zou, Y.; Wang, X.; Yin, C.; Pan, L.; Shen, Y.; Jia, J.; et al. Low density lipoprotein receptor related protein 6 (LRP6) protects heart against oxidative stress by the crosstalk of HSF1 and GSK3beta. Redox Biol. 2020, 37, 101699. [Google Scholar] [CrossRef]
- Kandilis, A.N.; Karidis, N.P.; Kouraklis, G.; Patsouris, E.; Vasileiou, I.; Theocharis, S. Proteasome inhibitors: Possible novel therapeutic strategy for ischemia-reperfusion injury? Expert Opin. Investig. Drugs 2014, 23, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Kukan, M. Emerging roles of proteasomes in ischemia-reperfusion injury of organs. J. Physiol. Pharmacol. 2004, 55 Pt 1, 3–15. [Google Scholar] [PubMed]
- Bao, J.; Sato, K.; Li, M.; Gao, Y.; Abid, R.; Aird, W.; Simons, M.; Post, M.J. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2612–H2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flohé, L.; Brigelius-Flohé, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox Regulation of NF-kappa B Activation. Free Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Li, D.; Pi, W.; Sun, Z.; Liu, X.; Jiang, J. Ferroptosis and its role in cardiomyopathy. Biomed. Pharmacother. 2022, 153, 113279. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Rafiq, K. Proteasome biology and therapeutics in cardiac diseases. Transl. Res. 2019, 205, 64–76. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, D.; Dong, X.; Zhang, X.; Liu, J.; Peng, L.; Meng, B.; Hua, Q.; Pei, X.; Zhao, L.; et al. LncDACH1 promotes mi-tochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci. China Life Sci. 2022, 65, 1198–1212. [Google Scholar] [CrossRef]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin Proteasome Dysfunction in Human Hypertrophic and Dilated Cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Gilda, J.E.; Lai, X.; Witzmann, F.A.; Gomes, A.V. Delineation of Molecular Pathways Involved in Cardiomyopathies Caused by Troponin T Mutations. Mol. Cell. Proteom. 2016, 15, 1962–1981. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, K.; Terasaki, F.; Shimomura, H.; Tsukada, B.; Horii, T.; Isomura, T.; Suma, H.; Shibayama, Y.; Kitaura, Y. Enhanced expression of the ubiquitin-proteasome system in the myocardium from patients with dilated cardiomyopathy referred for left ventriculoplasty: An immunohistochemical study with special reference to oxidative stress. Heart Vessel. 2010, 25, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Dvornikov, A.V.; Smolin, N.; Zhang, M.; Martin, J.L.; Robia, S.L.; de Tombe, P.P. Restrictive Cardiomyopathy Troponin I R145W Mutation Does Not Perturb Myofilament Length-dependent Activation in Human Cardiac Sarcomeres. J. Biol. Chem. 2016, 291, 21817–21828. [Google Scholar] [CrossRef] [PubMed]
- Goto, J.; Otaki, Y.; Watanabe, T.; Kobayashi, Y.; Aono, T.; Watanabe, K.; Wanezaki, M.; Kutsuzawa, D.; Kato, S.; Tamura, H.; et al. HECT (Homologous to the E6-AP Carboxyl Terminus)-Type Ubiquitin E3 Ligase ITCH Attenuates Cardiac Hypertrophy by Suppressing the Wnt/beta-Catenin Signaling Pathway. Hypertension 2020, 76, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Dadson, K.; Hauck, L.; Hao, Z.; Grothe, D.; Rao, V.; Mak, T.W.; Billia, F. The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1. Sci. Rep. 2017, 7, 41490. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Tang, Y.; Wang, J.; Lu, G.; Niu, J.; Wang, J.; Li, J.; Liu, Q.; Wang, Z.; Huang, Z.; et al. A new FGF1 variant protects against adriamycin-induced cardiotoxicity via modulating p53 activity. Redox Biol. 2022, 49, 102219. [Google Scholar] [CrossRef]
- Saettini, F.; Poli, C.; Vengoechea, J.; Bonanomi, S.; Orellana, J.C.; Fazio, G.; Rodriguez, F.H.; Noguera, L.P.; Booth, C.; Jarur-Chamy, V.; et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin-interacting protein 1 deficiency. Blood 2021, 137, 493–499. [Google Scholar] [CrossRef]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martínez-González, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodríguez-Pérez, F.; et al. Structural basis and regulation of the reductive stress response. Cell 2021, 184, 5375–5390.e16. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef] [Green Version]
- Manford, A.G.; Rodríguez-Pérez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61.e21. [Google Scholar] [CrossRef]
- Yu, W.; Chen, C.; Cheng, J. The role and molecular mechanism of FoxO1 in mediating cardiac hypertrophy. ESC Heart Fail. 2020, 7, 3497–3504. [Google Scholar] [CrossRef]
- Ali, A.; Kuo, W.W.; Kuo, C.H.; Lo, J.F.; Chen, M.Y.C.; Daddam, J.R.; Ho, T.J.; Viswanadha, V.P.; Shibu, M.A.; Huang, C.Y. E3 ligase activity of Carboxyl terminus of Hsc70 interacting protein (CHIP) in Wharton’s jelly derived mesenchymal stem cells improves their persistence under hyperglycemic stress and promotes the prophylactic effects against diabetic cardiac damages. Bioeng. Transl. Med. 2021, 6, e10234. [Google Scholar] [CrossRef] [PubMed]
- Zolk, O.; Schenke, C.; Sarikas, A. The ubiquitin–proteasome system: Focus on the heart. Cardiovasc. Res. 2006, 70, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, S.; Garbade, J.; Emrich, F.; Klaeske, K.; Borger, M.A.; Lehmann, S.; Jawad, K.; Dieterlen, M.T. Ischemic Cardio-myopathy Affects the Thioredoxin System in the Human Myocardium. J. Card. Fail. 2019, 25, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, W.; Deng, K.-Q.; Tian, S.; Liu, H.; Shi, H.; Fang, Q.; Liu, Z.; Chen, Z.; Tian, T.; et al. The E3 Ligase TRIM16 Is a Key Suppressor of Pathological Cardiac Hypertrophy. Circ. Res. 2022, 130, 1586–1600. [Google Scholar] [CrossRef]
- Hou, K.; Shen, J.; Yan, J.; Zhai, C.; Zhang, J.; Pan, J.-A.; Zhang, Y.; Jiang, Y.; Wang, Y.; Lin, R.Z.; et al. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. eBioMedicine 2021, 69, 103456. [Google Scholar] [CrossRef]
- Binder, P.; Nguyen, B.; Collins, L.; Zi, M.; Liu, W.; Christou, F.; Luo, X.; Hille, S.S.; Frey, N.; Cartwright, E.J.; et al. Pak2 Reg-ulation of Nrf2 Serves as a Novel Signaling Nexus Linking ER Stress Response and Oxidative Stress in the Heart. Front. Car-Diovasc Med. 2022, 9, 851419. [Google Scholar] [CrossRef]
- Qi, Y.; Tang, Y.; Yin, L.; Ding, K.; Zhao, C.; Yan, W.; Yao, Y. miR-129-5p restores cardiac function in rats with chronic heart failure by targeting the E3 ubiquitin ligase Smurf1 and promoting PTEN expression. Bioengineered 2022, 13, 2371–2386. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, W.; Bai, X.; Wang, X.; Wang, Y.; Yin, Y. microRNA-454-mediated NEDD4-2/TrkA/cAMP axis in heart failure: Mechanisms and cardioprotective implications. J. Cell. Mol. Med. 2021, 25, 5082–5098. [Google Scholar] [CrossRef]
- Bozi, L.H.; Jannig, P.R.; Rolim, N.; Voltarelli, V.A.; Dourado, P.M.; Wisløff, U.; Brum, P.C. Aerobic exercise training rescues cardiac protein quality control and blunts endoplasmic reticulum stress in heart failure rats. J. Cell. Mol. Med. 2016, 20, 2208–2212. [Google Scholar] [CrossRef] [Green Version]
- Mangner, N.; Bowen, T.S.; Werner, S.; Fischer, T.; Kullnick, Y.; Oberbach, A.; Linke, A.; Steil, L.; Schuler, G.; Adams, V. Exercise Training Prevents Diaphragm Contractile Dysfunction in Heart Failure. Med. Sci. Sports Exerc. 2016, 48, 2118–2124. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhang, F.; Yan, P.; Zhang, S.; Lou, Y.; Geng, Z.; Li, Z.; Zhang, Y.; Xu, Y.; Lu, Y.; et al. LARP7 Protects Against Heart Failure by Enhancing Mitochondrial Biogenesis. Circulation 2021, 143, 2007–2022. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, S.; Asahi, M. Phospholamban Is Downregulated by pVHL-Mediated Degradation through Oxidative Stress in Failing Heart. Int. J. Mol. Sci. 2017, 18, 2232. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Nicholson, C.K.; Lambert, J.P.; Barr, L.A.; Kuek, N.; Herszenhaut, D.; Tan, L.; Murohara, T.; Hansen, J.M.; Husain, A.; et al. Sodium Sulfide Attenuates Ischemic-Induced Heart Failure by Enhancing Proteasomal Function in an Nrf2-Dependent Manner. Circ. Heart Fail. 2016, 9, e002368. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, M.; Chen, J.; Li, X.; Zhuang, J. Intersection of the Ubiquitin–Proteasome System with Oxidative Stress in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 12197. https://doi.org/10.3390/ijms232012197
Qiu M, Chen J, Li X, Zhuang J. Intersection of the Ubiquitin–Proteasome System with Oxidative Stress in Cardiovascular Disease. International Journal of Molecular Sciences. 2022; 23(20):12197. https://doi.org/10.3390/ijms232012197
Chicago/Turabian StyleQiu, Min, Jimei Chen, Xiaohong Li, and Jian Zhuang. 2022. "Intersection of the Ubiquitin–Proteasome System with Oxidative Stress in Cardiovascular Disease" International Journal of Molecular Sciences 23, no. 20: 12197. https://doi.org/10.3390/ijms232012197
APA StyleQiu, M., Chen, J., Li, X., & Zhuang, J. (2022). Intersection of the Ubiquitin–Proteasome System with Oxidative Stress in Cardiovascular Disease. International Journal of Molecular Sciences, 23(20), 12197. https://doi.org/10.3390/ijms232012197